
 Technology peripheralsAIWorld's first: Molecular Heart's new AI algorithm to overcome the problems of protein side chain prediction and sequence design
Technology peripheralsAIWorld's first: Molecular Heart's new AI algorithm to overcome the problems of protein side chain prediction and sequence design
Heart of Machine Column
Heart of Machine Editorial Department
PSCP deep architecture AttnPacker - greatly optimized AI algorithm.
The formation of protein structure and function depends largely on the interaction between side chain atoms. Therefore, accurate protein side chain prediction (PSCP) is a key link in solving the problems of protein structure prediction and protein design. However, previous protein structure predictions mostly focused on the main chain structure, and side chain structure prediction has always been a difficult problem that has not been completely solved.
Recently, Xu Jinbo’s team at Molecular Heart launched a new PSCP deep architecture AttnPacker, which has achieved significant improvements in speed, memory efficiency and overall accuracy. It is currently the best known side-chain structure prediction algorithm and the first in the world. An AI algorithm that can perform protein side chain prediction and sequence design at the same time.
The paper was published in the Proceedings of the National Academy of Sciences (PNAS), and its pre-training model, source code and inference scripts have been open sourced on Github.

Paper link:
https://www.pnas.org/doi/10.1073/pnas.2216438120#supplementary-materials
Open source link:
https://github.com/MattMcPartlon/AttnPacker
background
Protein is folded from several amino acids, and its structure is divided into main chain and side chain. Differences in side chains have a huge impact on protein structure and function, especially biological activity. Based on a clear understanding of the side chain structure, scientists can more accurately determine the three-dimensional structure of proteins, analyze protein-protein interactions, and conduct rational protein design. When applied to the field of drug design, scientists can quickly and more accurately find suitable binding sites for drugs and receptors, and even optimize or design binding sites as needed; in the field of enzyme optimization, scientists can optimize sequences Transformation allows multiple side chains to participate in catalytic reactions to achieve more efficient and specific catalytic effects.
Most of the current protein structure prediction algorithms mainly focus on the structural analysis of the main chain, but protein side chain structure prediction is still a problem that has not been completely overcome. Whether it is popular protein structure prediction algorithms such as AlphaFold2 or algorithms focusing on side chain structure prediction such as DLPacker and RosettaPacker, the accuracy or speed are not satisfactory. This also imposes limitations on protein design.
Traditional methods, such as RosettaPacker, mainly use energy optimization methods, first grouping the distribution of side chain atoms, and then searching the grouping of side chains for a specific amino acid to find the combination with the smallest energy. These methods differ primarily from the researcher's choice of rotamer libraries, energy functions, and energy minimization procedures, with accuracy limited by the use of search heuristics and discrete sampling procedures. There are also side chain prediction methods based on deep learning in the industry, such as DLPacker, which formulates PSCP as an image-to-image conversion problem and adopts a U-net model structure. However, the prediction accuracy and speed are still not ideal.
method
AttnPacker is an end-to-end deep learning method for predicting protein side chain coordinates. It jointly simulates side chain interactions, with directly predicted side chain structures that are more physically feasible, with fewer atomic collisions and more ideal bond lengths and angles.
Specifically, AttnPacker introduces a depth map converter architecture that leverages the geometric and relational aspects of PSCP. Inspired by AlphaFold2, Molecular Heart proposes position-aware triangle updates to optimize pairwise features using a graph-based framework to compute triangle attention and multiplicative updates. With this approach, AttnPacker has significantly less memory and a higher capacity model. Furthermore, Molecular Heart explores several SE (3) equivariant attention mechanisms and proposes an equivariant transformer architecture for learning from 3D points.

AttnPacker runs the process. The protein backbone coordinates and sequence are used as input, and the spatial feature map and equivariable basis are derived based on the coordinate information. The feature map is processed by the invariant graph-transformer module and then passed to an equivariant TFN-Transformer that outputs predicted side chain coordinates, confidence scores for each residue, and optional design sequences. The predicted coordinates are post-processed to remove all spatial conflicts and ensure idealized geometry.
Effect
In terms of prediction performance, AttnPacker shows improvements in accuracy and efficiency for both natural and non-natural backbone structures. At the same time, physical feasibility is ensured, deviations from ideal bond lengths and angles are negligible, and minimal atomic steric hindrance is produced.
Molecular Heart conducted a comparative test on AttnPacker with the current state-of-the-art methods - SCWRL4, FASPR, RosettaPacker and DLPacker on the CASP13 and CASP14 natural and non-native protein backbone data sets. Results show that AttnPacker significantly outperforms traditional protein side chain prediction methods on CASP13 and CASP14 native backbones, with average reconstruction RMSDs more than 18% lower than the suboptimal method on each test set. AttnPacker also outperforms the deep learning method DLPacker, reducing average RMSD by more than 11% while also significantly improving sidechain dihedral accuracy. In addition to accuracy, AttnPacker has significantly fewer atomic collisions than other methods.

Given the natural main chain structure, the side chain structure prediction results of each algorithm on the CASP13 and CASP14 target proteins are given. Asterisks indicate that the average conflict values are lower than the native structure—56.0, 5.9, and 0.4 for CASP13 and 80.4, 7.9, and 2.5 for CASP14.
On CASP13 and CASP14 non-native backbones, AttnPacker is also significantly better than other methods, and atomic collisions are also significantly less than other methods.

Given the non-natural backbone structure, the side chain structure prediction results of each algorithm on the CASP13 and CASP14 target proteins are given. Asterisks indicate that the average conflict values are lower than the corresponding native structures—34.6, 2.2, 0.5 for CASP13 and 40.0, 2.7, 0.7 for CASP14.
Innovatively abandons the discrete rotamer library and computationally expensive conformational search and sampling steps, and directly combines the main chain 3D geometric structure to calculate all side chain coordinates in parallel. Compared with the deep learning-based method DLPacker and the traditional computing method-based RosettaPacker, AttnPacker has significantly improved computing efficiency and reduced inference time by more than 100 times.

Time comparison of different PSCP methods. Reconstructing the relative times of side chain atoms for all 83 CASP13 target proteins.
AttnPacker performs equally well in protein design. Molecular Heart trained an AttnPacker variant for co-design that achieves native sequence recovery rates comparable to current state-of-the-art methods while also producing highly accurate assemblies. Rosetta simulation validation shows that AttnPacker-designed structures generally produce subnative (lower) Rosetta energies.

The ESMFold scTM and plDDT indicators were used to compare the native protein sequence and the sequence generated by AttnPacker to evaluate the quality of AttnPacker's generation, and the results showed a strong correlation.
In addition to its amazing effects and efficiency, AttnPaker also has a very practical value - it is very easy to use. AttnPaker only requires a protein structure file to run. In contrast, OPUS-Rota4 (28) requires a voxel representation of the atomic environment from DLPacker, logic, secondary structure from trRosetta100, and constraint files from OPUS-CM output. Additionally, since AttnPacker directly predicts side chain coordinates, the output is fully differentiable, which facilitates downstream prediction tasks such as optimization or protein-protein interactions. "The advantages of good prediction effect, high efficiency and ease of use are conducive to the widespread use of AttnPacker in research and industrial fields." Professor Xu Jinbo said.
Summarize
1. AttnPacker is a SE (3) equivariant model used to directly predict sequence and side chain coordinates. It can be used for protein side chain structure prediction and protein sequence design. It is a pioneering work.
2. AttnPacker's accuracy is better than other methods, its efficiency is greatly improved, and it is extremely easy to use.
The above is the detailed content of World's first: Molecular Heart's new AI algorithm to overcome the problems of protein side chain prediction and sequence design. For more information, please follow other related articles on the PHP Chinese website!

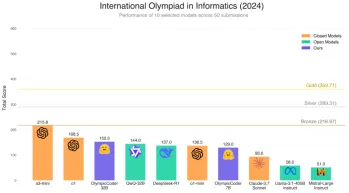 Does Hugging Face's 7B Model OlympicCoder Beat Claude 3.7?Apr 23, 2025 am 11:49 AM
Does Hugging Face's 7B Model OlympicCoder Beat Claude 3.7?Apr 23, 2025 am 11:49 AMHugging Face's OlympicCoder-7B: A Powerful Open-Source Code Reasoning Model The race to develop superior code-focused language models is intensifying, and Hugging Face has joined the competition with a formidable contender: OlympicCoder-7B, a product
 4 New Gemini Features You Can't Afford to MissApr 23, 2025 am 11:48 AM
4 New Gemini Features You Can't Afford to MissApr 23, 2025 am 11:48 AMHow many of you have wished AI could do more than just answer questions? I know I have, and as of late, I’m amazed by how it’s transforming. AI chatbots aren’t just about chatting anymore, they’re about creating, researchin
 Camunda Writes New Score For Agentic AI OrchestrationApr 23, 2025 am 11:46 AM
Camunda Writes New Score For Agentic AI OrchestrationApr 23, 2025 am 11:46 AMAs smart AI begins to be integrated into all levels of enterprise software platforms and applications (we must emphasize that there are both powerful core tools and some less reliable simulation tools), we need a new set of infrastructure capabilities to manage these agents. Camunda, a process orchestration company based in Berlin, Germany, believes it can help smart AI play its due role and align with accurate business goals and rules in the new digital workplace. The company currently offers intelligent orchestration capabilities designed to help organizations model, deploy and manage AI agents. From a practical software engineering perspective, what does this mean? The integration of certainty and non-deterministic processes The company said the key is to allow users (usually data scientists, software)
 Is There Value In A Curated Enterprise AI Experience?Apr 23, 2025 am 11:45 AM
Is There Value In A Curated Enterprise AI Experience?Apr 23, 2025 am 11:45 AMAttending Google Cloud Next '25, I was keen to see how Google would distinguish its AI offerings. Recent announcements regarding Agentspace (discussed here) and the Customer Experience Suite (discussed here) were promising, emphasizing business valu
 How to Find the Best Multilingual Embedding Model for Your RAG?Apr 23, 2025 am 11:44 AM
How to Find the Best Multilingual Embedding Model for Your RAG?Apr 23, 2025 am 11:44 AMSelecting the Optimal Multilingual Embedding Model for Your Retrieval Augmented Generation (RAG) System In today's interconnected world, building effective multilingual AI systems is paramount. Robust multilingual embedding models are crucial for Re
 Musk: Robotaxis In Austin Need Intervention Every 10,000 MilesApr 23, 2025 am 11:42 AM
Musk: Robotaxis In Austin Need Intervention Every 10,000 MilesApr 23, 2025 am 11:42 AMTesla's Austin Robotaxi Launch: A Closer Look at Musk's Claims Elon Musk recently announced Tesla's upcoming robotaxi launch in Austin, Texas, initially deploying a small fleet of 10-20 vehicles for safety reasons, with plans for rapid expansion. H
 AI's Shocking Pivot: From Work Tool To Digital Therapist And Life CoachApr 23, 2025 am 11:41 AM
AI's Shocking Pivot: From Work Tool To Digital Therapist And Life CoachApr 23, 2025 am 11:41 AMThe way artificial intelligence is applied may be unexpected. Initially, many of us might think it was mainly used for creative and technical tasks, such as writing code and creating content. However, a recent survey reported by Harvard Business Review shows that this is not the case. Most users seek artificial intelligence not just for work, but for support, organization, and even friendship! The report said that the first of AI application cases is treatment and companionship. This shows that its 24/7 availability and the ability to provide anonymous, honest advice and feedback are of great value. On the other hand, marketing tasks (such as writing a blog, creating social media posts, or advertising copy) rank much lower on the popular use list. Why is this? Let's see the results of the research and how it continues to be
 Companies Race Toward AI Agent AdoptionApr 23, 2025 am 11:40 AM
Companies Race Toward AI Agent AdoptionApr 23, 2025 am 11:40 AMThe rise of AI agents is transforming the business landscape. Compared to the cloud revolution, the impact of AI agents is predicted to be exponentially greater, promising to revolutionize knowledge work. The ability to simulate human decision-maki


Hot AI Tools

Undresser.AI Undress
AI-powered app for creating realistic nude photos

AI Clothes Remover
Online AI tool for removing clothes from photos.

Undress AI Tool
Undress images for free

Clothoff.io
AI clothes remover

Video Face Swap
Swap faces in any video effortlessly with our completely free AI face swap tool!

Hot Article
Hot Tools

Dreamweaver CS6
Visual web development tools

MantisBT
Mantis is an easy-to-deploy web-based defect tracking tool designed to aid in product defect tracking. It requires PHP, MySQL and a web server. Check out our demo and hosting services.

SublimeText3 Mac version
God-level code editing software (SublimeText3)

VSCode Windows 64-bit Download
A free and powerful IDE editor launched by Microsoft

SublimeText3 Chinese version
Chinese version, very easy to use

