



根據外媒Tech Xplore報道,麻省理工學院的研究人員最近開發了一種叫做EquBind的新模型,這個模型可以提前預測新蛋白質分子的結構,提升藥物開發的效率。
目前這項技術已經得到了業界內的認可,闡述這項技術的論文也將在7月被國際機器學習會議(ICML)會議接收。
一、速度提升1200倍,EquBind模型能迅速篩選類藥物分子
目前,藥物研發是一件漫長而又昂貴的事情。其中最主要的原因就是開發藥物的成本十分昂貴。這種成本不僅包括數十億美元的資金投入,還包括長達數十年的研究時間。
而且在研發的過程中,90%的藥物都會因為無效或副作用太多而研發失敗,只有10%的藥物能夠順利通過食品藥物管理局的檢查,被批准上市。
因此,藥廠會提高研發成功藥品的價格,來彌補研發失敗藥品造成的損失,所以目前有些藥物的價格居高不下。

▲一些蛋白質分子結構
如果研究人員想要進行藥物開發,就要先找到有開發潛力的類藥物分子(drug-like molecules)。藥物研發進展緩慢還有另一個重要的原因,那就是現存的類藥物分子數量龐大。數據顯示,目前現存的類藥物分子多達1016種,這個數字遠遠超出了現有的分子計算模型的計算上限。
為了處理資料如此龐大的分子,加快藥物開發的進程,麻省理工學院電子工程和電腦科學系的研一學生Hannes St rk開發了一種稱為「EquBind」的幾何深度學習模型。 EquBind比現存最快的分子計算對接模型運行速度快1200倍,能夠更快找到類藥物分子。
二、EquBind模型能精準預測蛋白質結構,提升藥物研發效率
目前大多數傳統的分子計算對接模型都是透過一種稱為「配體-蛋白質」(ligand- to-protein binding)的方法尋找類藥物分子。具體而言,模型需要先接收大量的樣本分子,然後讓配體與各種分子結合,然後模型再對不同分子進行評分,再以最後的排名來篩選出最合適的分子。但這種做法流程繁複,模型尋找類藥物分子的效率較低。
Hannes St rk對這個過程做了一個形象的比喻,他說:「以前的典型的『配體-蛋白質』方法就好像是試圖讓模型把鑰匙插入有很多鑰匙孔的鎖,模型要花費大量的時間為鑰匙和每一個鎖孔的適配度打分,再選出最合適的那個。」
他繼續解釋:「而EquBind可以跳過最花費時間的步驟,可以在遇到新分子時提前預測最合適的'鎖孔',這就是所謂的'盲配對'(blind docking)。EquBind有內置的幾何推理算法,能夠幫助模型學習分子的基本結構。這個算法可以讓EquBind在遇到新的分子時直接預測出最合適的位置,而不用花費大量的時間嘗試不同的位置並打分數。」

▲麻省理工學院
三、EquBind模型已在業界成功應用,作者期待更多反饋
這個模型引起了治療公司Relay的首席數據官帕特·沃爾特斯(Pat Walters)的注意。沃爾斯特建議Hannes St rk的研究團隊使用這種模型來進行用於治療肺癌、白血病和胃腸道腫瘤的藥物開發。通常而言,用於這些領域藥物的蛋白質配體很難用大多數傳統的方法對接,但是EquBind卻能讓它們成功對接。

▲兩種治療肺癌的抑制劑藥物
沃特斯說:「EquBind為蛋白質對接問題提供了一種獨特的解決方案,它解決了結構預測和綁定位點識別等問題。這種方法可以很好地利用數千種公開的晶體結構信息,EquBind可能會以新的方式影響這個領域。」
發表這項技術的論文將在7月被國際機器學習會議(ICML)接收,該論文的作者Hannes St rk表示:「我很期待能在這次會議上收到一些關於EquBind模型的改進意見。」
結論:AI與製藥適配度極佳,發展勢頭正盛
AI製藥是一個2020年才走進公眾視野的新興領域。
製藥領域是一個天然的AI場景。新藥研發的長週期、高成本、低成功率,為AI留下了龐大的用武之地:機器可以自主學習數據、挖掘數據,總結歸納專家經驗外的藥物研發規律,繼而優化藥物研發流程中的各環節,不僅可以提升藥物研發效率與成功率,還有望降低研發費用與試誤成本。
因為這樣的特性和發展潛力,目前AI製藥勢頭正盛。但也有業內人士唱衰,說AI在製藥過程中扮演的終歸只是輔助角色,繞不開行業固有的流程和機制,不可能用兩三年的時間做完十年的事。
但整體而言,目前AI製藥領域還是不斷有新的技術突破,發展蒸蒸日上。
以上是效率提升1200倍!麻省理工開發AI製藥新模型的詳細內容。更多資訊請關注PHP中文網其他相關文章!

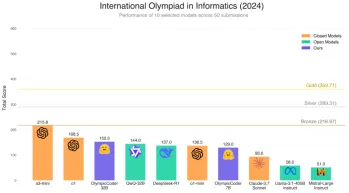 擁抱面部是否7B型號奧林匹克賽車擊敗克勞德3.7?Apr 23, 2025 am 11:49 AM
擁抱面部是否7B型號奧林匹克賽車擊敗克勞德3.7?Apr 23, 2025 am 11:49 AM擁抱Face的OlympicCoder-7B:強大的開源代碼推理模型 開發以代碼為中心的語言模型的競賽正在加劇,擁抱面孔與強大的競爭者一起參加了比賽:OlympicCoder-7B,一種產品
 4個新的雙子座功能您可以錯過Apr 23, 2025 am 11:48 AM
4個新的雙子座功能您可以錯過Apr 23, 2025 am 11:48 AM你們當中有多少人希望AI可以做更多的事情,而不僅僅是回答問題?我知道我有,最近,我對它的變化感到驚訝。 AI聊天機器人不僅要聊天,還關心創建,研究
 Camunda為經紀人AI編排編寫了新的分數Apr 23, 2025 am 11:46 AM
Camunda為經紀人AI編排編寫了新的分數Apr 23, 2025 am 11:46 AM隨著智能AI開始融入企業軟件平台和應用程序的各個層面(我們必須強調的是,既有強大的核心工具,也有一些不太可靠的模擬工具),我們需要一套新的基礎設施能力來管理這些智能體。 總部位於德國柏林的流程編排公司Camunda認為,它可以幫助智能AI發揮其應有的作用,並與新的數字工作場所中的準確業務目標和規則保持一致。該公司目前提供智能編排功能,旨在幫助組織建模、部署和管理AI智能體。 從實際的軟件工程角度來看,這意味著什麼? 確定性與非確定性流程的融合 該公司表示,關鍵在於允許用戶(通常是數據科學家、軟件
 策劃的企業AI體驗是否有價值?Apr 23, 2025 am 11:45 AM
策劃的企業AI體驗是否有價值?Apr 23, 2025 am 11:45 AM參加Google Cloud Next '25,我渴望看到Google如何區分其AI產品。 有關代理空間(此處討論)和客戶體驗套件(此處討論)的最新公告很有希望,強調了商業價值
 如何為抹布找到最佳的多語言嵌入模型?Apr 23, 2025 am 11:44 AM
如何為抹布找到最佳的多語言嵌入模型?Apr 23, 2025 am 11:44 AM為您的檢索增強發電(RAG)系統選擇最佳的多語言嵌入模型 在當今的相互聯繫的世界中,建立有效的多語言AI系統至關重要。 強大的多語言嵌入模型對於RE至關重要
 麝香:奧斯汀的機器人需要每10,000英里進行干預Apr 23, 2025 am 11:42 AM
麝香:奧斯汀的機器人需要每10,000英里進行干預Apr 23, 2025 am 11:42 AM特斯拉的Austin Robotaxi發射:仔細觀察Musk的主張 埃隆·馬斯克(Elon Musk)最近宣布,特斯拉即將在德克薩斯州奧斯汀推出的Robotaxi發射,最初出於安全原因部署了一支小型10-20輛汽車,並有快速擴張的計劃。 h
 AI震驚的樞軸:從工作工具到數字治療師和生活教練Apr 23, 2025 am 11:41 AM
AI震驚的樞軸:從工作工具到數字治療師和生活教練Apr 23, 2025 am 11:41 AM人工智能的應用方式可能出乎意料。最初,我們很多人可能認為它主要用於代勞創意和技術任務,例如編寫代碼和創作內容。 然而,哈佛商業評論最近報導的一項調查表明情況並非如此。大多數用戶尋求人工智能的並非是代勞工作,而是支持、組織,甚至是友誼! 報告稱,人工智能應用案例的首位是治療和陪伴。這表明其全天候可用性以及提供匿名、誠實建議和反饋的能力非常有價值。 另一方面,營銷任務(例如撰寫博客、創建社交媒體帖子或廣告文案)在流行用途列表中的排名要低得多。 這是為什麼呢?讓我們看看研究結果及其對我們人類如何繼續將



熱AI工具

Undresser.AI Undress
人工智慧驅動的應用程序,用於創建逼真的裸體照片

AI Clothes Remover
用於從照片中去除衣服的線上人工智慧工具。

Undress AI Tool
免費脫衣圖片

Clothoff.io
AI脫衣器

Video Face Swap
使用我們完全免費的人工智慧換臉工具,輕鬆在任何影片中換臉!

熱門文章
熱工具

mPDF
mPDF是一個PHP庫,可以從UTF-8編碼的HTML產生PDF檔案。原作者Ian Back編寫mPDF以從他的網站上「即時」輸出PDF文件,並處理不同的語言。與原始腳本如HTML2FPDF相比,它的速度較慢,並且在使用Unicode字體時產生的檔案較大,但支援CSS樣式等,並進行了大量增強。支援幾乎所有語言,包括RTL(阿拉伯語和希伯來語)和CJK(中日韓)。支援嵌套的區塊級元素(如P、DIV),

VSCode Windows 64位元 下載
微軟推出的免費、功能強大的一款IDE編輯器

記事本++7.3.1
好用且免費的程式碼編輯器

PhpStorm Mac 版本
最新(2018.2.1 )專業的PHP整合開發工具

ZendStudio 13.5.1 Mac
強大的PHP整合開發環境

