



重新编写的内容是:紫罗
复杂电子相互作用的大规模模拟仍然是原子建模面临的最大挑战之一。虽然经典力场通常无法描述电子态和离子重排之间的耦合,但更准确的从头算分子动力学受到计算复杂性的影响,无法进行长时间和大规模的模拟,而这对于研究技术相关现象至关重要
近日,来自加州大学伯克利分校和劳伦斯伯克利国家实验室的研究人员,提出了一种基于图神经网络的机器学习原子间势(MLIP)模型:晶体哈密顿图神经网络(Crystal Hamiltonian Graph Neural Network,CHGNet),可以对通用势能面进行建模。
研究强调了电荷信息对于捕获适当的化学反应的重要性,并提供了对离子系统的见解,这些离子系统具有以前的 MLIP 无法观察到的额外电子自由度。
该研究以「CHGNet as a pretrained universal neural network potential for charge-informed atomistic modelling」为题,于 2023 年 9 月 14 日发布在《Nature Machine Intelligence》上。

大规模模拟,如分子动力学(MD),是固态材料计算探索的重要工具。然而,精确建模电子相互作用及其在分子动力学模拟中微妙影响仍然是一个巨大的挑战。经典力场等经验方法通常不够准确,无法捕捉复杂的电子相互作用
从头算分子动力学 (AIMD) 与密度泛函理论 (DFT) 相结合,可以通过显式计算密度泛函近似内的电子结构,产生具有量子力学精度的高保真结果。长时间、大规模的自旋极化 AIMD 模拟对于研究离子迁移、相变和化学反应至关重要,具有挑战性且计算量极大。
诸如 ænet 和 DeepMD 之类的 MLIP,为弥合昂贵的电子结构方法和高效的经典原子间电势之间的差距提供了有希望的解决方案。然而,包含化合价对化学键的重要影响仍然是 MLIP 的一个挑战。
电荷可以通过多种方式来表示,从简单的氧化态标签到从量子力学推导出的连续波函数。将电荷信息纳入MLIP的挑战来自于许多因素,例如表示的模糊性、解释的复杂性、标签的稀缺性等
需要重新写作的内容是:CHGNet 架构
CHGNet根据材料项目轨迹数据集(MPtrj)的能量、力、应力和磁矩进行了预训练。该数据集包含了对150万个无机结构进行了10多年的密度泛函理论计算。通过明确包含磁矩,CHGNet能够学习并准确表示电子的轨道占据情况,从而增强了其描述原子和电子自由度的能力
MPtrj 数据集中元素的分布如下图所示
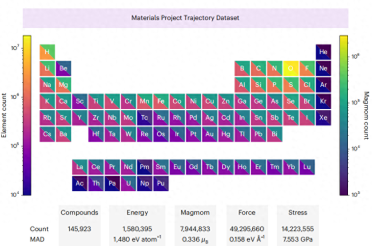
重写内容:图示:MPtrj数据集中元素的分布情况。(来源:论文)
在这里,研究人员将电荷定义为一种原子属性(原子电荷),可以通过包含磁矩(magmoms)来推断。研究表明,通过明确地将特定位点的 magmoms 作为电荷态约束纳入 CHGNet 中,既可以增强潜在空间正则化,又可以准确捕获电子相互作用
CHGNet 的基础是 GNN,其中图卷积层用于通过由边 {eij} 连接的一组节点 {vi} 传播原子信息。GNN 中保留了平移、旋转和排列不变性。CHGNet 以具有未知原子电荷的晶体结构作为输入,并输出相应的能量、力、应力和 magmoms。电荷装饰结构可以从现场 magmoms 和原子轨道理论推断出来。

重写内容如下:图示:CHGNet 模型架构。(来源:论文)
在 CHGNet 中,通过在原始单元中每个原子 vi 的 内搜索相邻原子 vj,将周期性晶体结构转换为原子图
内搜索相邻原子 vj,将周期性晶体结构转换为原子图 。
。
与其他 GNN 不同,其中 t 个卷积层后更新的原子特征 直接用于预测能量,CHGNet 正则化 t−1 卷积层的节点特征
直接用于预测能量,CHGNet 正则化 t−1 卷积层的节点特征
 以包含有关岩浆的信息。正则化特征
以包含有关岩浆的信息。正则化特征 携带有关局部离子环境和电荷分布的丰富信息。因此,用于预测能量、力和应力的原子特征
携带有关局部离子环境和电荷分布的丰富信息。因此,用于预测能量、力和应力的原子特征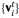 是受其电荷态信息约束的电荷。因此,CHGNet 可以仅使用核位置和原子身份作为输入来提供电荷态信息,从而可以研究原子建模中的电荷分布。
是受其电荷态信息约束的电荷。因此,CHGNet 可以仅使用核位置和原子身份作为输入来提供电荷态信息,从而可以研究原子建模中的电荷分布。
CHGNet 在固态材料中的应用
研究人员展示了 CHGNet 在固态材料中的几种应用。展示了 Na2V2(PO4)3 中原子电荷的电荷约束和潜在空间正则化,并展示了 CHGNet 在 LixMnO2 中的电荷转移和相变、LixFePO4 相图中的电子熵以及石榴石型锂超离子导体 Li3+xLa3Te2O12 中的锂 (Li) 和扩散率。
为了合理化对原子电荷的处理,使用 NASICON 型钠离子阴极材料 Na4V2(PO4)3 作为说明性示例。除了从 V 原子核的空间配位中学习之外,在没有任何有关 V 离子电荷分布的先验知识的情况下,CHGNet 成功地将 V 离子区分为三价 V 和四价 V 两组。

图中展示了Na2V2(PO4)3中的磁矩和隐藏空间的规范化。 (引用自论文)
在 LixFePO4 的研究中强调了 CHGNet 区分

的能力,这对于包含电子熵和有限温度相稳定性至关重要。
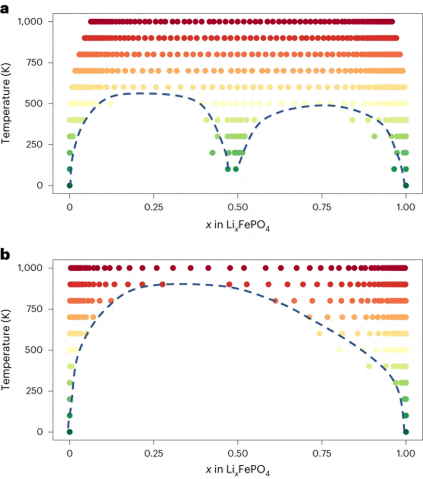
图示:来自 CHGNet 的 LixFePO4 相图。(来源:论文)
在 LiMnO2 的研究中,证明了 CHGNet 能够通过长时间的电荷信息 MD 深入了解异价过渡金属氧化物体系中电荷歧化和相变之间的关系。
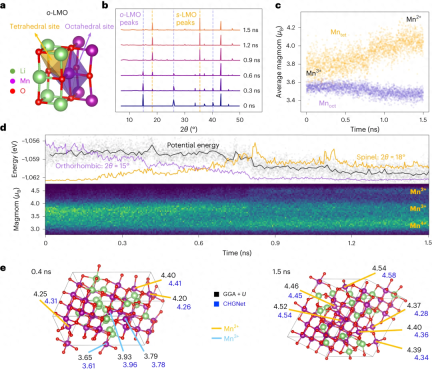
重写内容:图示:Li0.5MnO2的相变和电荷分化。(引自:论文)
接下来,我们研究了CHGNet在通用分子动力学模拟中的准确性。我们以石榴石导体中锂扩散为研究对象

图示:石榴石 Li3La3Te2O12 中的锂扩散率。(来源:论文)
结果显示,CHGNet不仅能够准确捕捉到活化扩散网络效应,而且其活化能量与DFT结果非常一致。这证明了CHGNet能够准确捕捉到锂离子在局部环境中的强相互作用,并具备模拟高度非线性扩散行为的能力。此外,CHGNet能够显著减少模拟扩散率的误差,并且通过扩展到纳秒级模拟,能够研究扩散率较差的系统
可进一步改进
虽然已经取得了以上的进步,但仍然有进一步改进的空间
首先,使用 magmom 进行价态推断并不能严格确保全局电荷中性
其次,尽管对于离子系统中自旋极化计算的原子电荷来说,magmom是一个很好的启发式方法,但人们意识到对于非磁性离子的原子电荷推断可能是不明确的,因此需要额外的领域知识。因此,对于没有magmom的离子,以原子为中心的magmom无法准确反映其原子电荷,CHGNet将从环境中推断电荷,类似于其他MLIP的功能
可以通过结合其他电荷表示方法来进一步增强模型,例如电子定位函数、电极化和基于原子轨道的划分。这些方法可以用于原子特征工程在潜在空间中
CHGNet 可以实现基于电荷的原子模拟,适用于大规模计算模拟以研究异价体系,从而扩大了计算化学、物理学、生物学和材料科学中电荷转移耦合现象的研究机会
请点击以下链接查看论文:https://www.nature.com/articles/s42256-023-00716-3
以上是基于电荷的原子模拟实现,利用预训练通用神经网络CHGNet的详细内容。更多信息请关注PHP中文网其他相关文章!

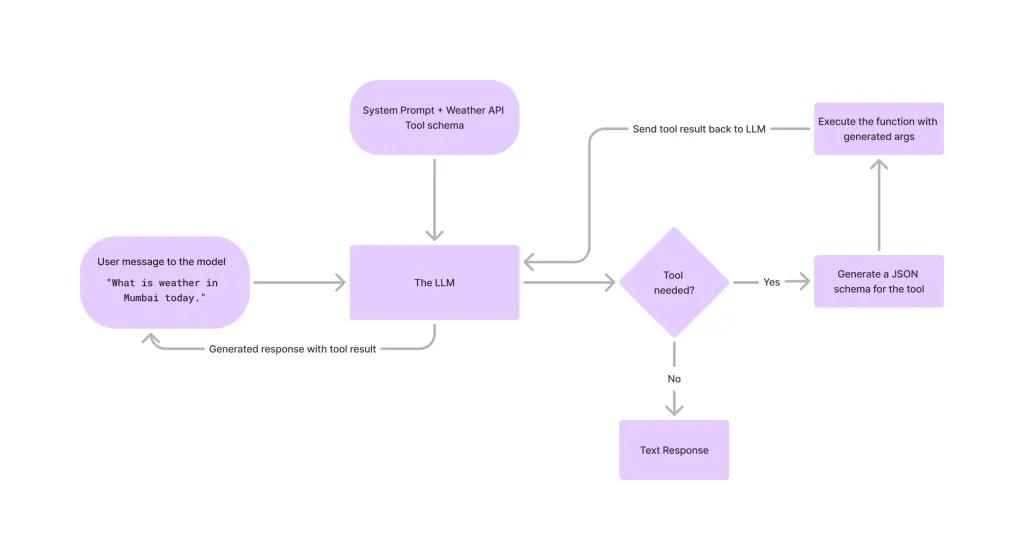 在LLMS中调用工具Apr 14, 2025 am 11:28 AM
在LLMS中调用工具Apr 14, 2025 am 11:28 AM大型语言模型(LLMS)的流行激增,工具称呼功能极大地扩展了其功能,而不是简单的文本生成。 现在,LLM可以处理复杂的自动化任务,例如Dynamic UI创建和自主a
 多动症游戏,健康工具和AI聊天机器人如何改变全球健康Apr 14, 2025 am 11:27 AM
多动症游戏,健康工具和AI聊天机器人如何改变全球健康Apr 14, 2025 am 11:27 AM视频游戏可以缓解焦虑,建立焦点或支持多动症的孩子吗? 随着医疗保健在全球范围内挑战,尤其是在青年中的挑战,创新者正在转向一种不太可能的工具:视频游戏。现在是世界上最大的娱乐印度河之一
 没有关于AI的投入:获胜者,失败者和机遇Apr 14, 2025 am 11:25 AM
没有关于AI的投入:获胜者,失败者和机遇Apr 14, 2025 am 11:25 AM“历史表明,尽管技术进步推动了经济增长,但它并不能自行确保公平的收入分配或促进包容性人类发展,”乌托德秘书长Rebeca Grynspan在序言中写道。
 通过生成AI学习谈判技巧Apr 14, 2025 am 11:23 AM
通过生成AI学习谈判技巧Apr 14, 2025 am 11:23 AM易于使用,使用生成的AI作为您的谈判导师和陪练伙伴。 让我们来谈谈。 对创新AI突破的这种分析是我正在进行的《福布斯》列的最新覆盖范围的一部分,包括识别和解释
 泰德(Ted)从Openai,Google,Meta透露出庭,与我自己自拍Apr 14, 2025 am 11:22 AM
泰德(Ted)从Openai,Google,Meta透露出庭,与我自己自拍Apr 14, 2025 am 11:22 AM在温哥华举行的TED2025会议昨天在4月11日举行了第36版。它有来自60多个国家 /地区的80个发言人,包括Sam Altman,Eric Schmidt和Palmer Luckey。泰德(Ted)的主题“人类重新构想”是量身定制的
 约瑟夫·斯蒂格利兹(Joseph StiglitzApr 14, 2025 am 11:21 AM
约瑟夫·斯蒂格利兹(Joseph StiglitzApr 14, 2025 am 11:21 AM约瑟夫·斯蒂格利茨(Joseph Stiglitz)是2001年著名的经济学家,是诺贝尔经济奖的获得者。斯蒂格利茨认为,AI可能会使现有的不平等和合并权力恶化,并在几个主导公司的手中加剧,最终破坏了经济的经济。
 什么是图形数据库?Apr 14, 2025 am 11:19 AM
什么是图形数据库?Apr 14, 2025 am 11:19 AM图数据库:通过关系彻底改变数据管理 随着数据的扩展及其特征在各个字段中的发展,图形数据库正在作为管理互连数据的变革解决方案的出现。与传统不同
 LLM路由:策略,技术和Python实施Apr 14, 2025 am 11:14 AM
LLM路由:策略,技术和Python实施Apr 14, 2025 am 11:14 AM大型语言模型(LLM)路由:通过智能任务分配优化性能 LLM的快速发展的景观呈现出各种各样的模型,每个模型都具有独特的优势和劣势。 有些在创意内容gen上表现出色


热AI工具

Undresser.AI Undress
人工智能驱动的应用程序,用于创建逼真的裸体照片

AI Clothes Remover
用于从照片中去除衣服的在线人工智能工具。

Undress AI Tool
免费脱衣服图片

Clothoff.io
AI脱衣机

AI Hentai Generator
免费生成ai无尽的。

热门文章
热工具

SublimeText3汉化版
中文版,非常好用

SublimeText3 Mac版
神级代码编辑软件(SublimeText3)

适用于 Eclipse 的 SAP NetWeaver 服务器适配器
将Eclipse与SAP NetWeaver应用服务器集成。

EditPlus 中文破解版
体积小,语法高亮,不支持代码提示功能

DVWA
Damn Vulnerable Web App (DVWA) 是一个PHP/MySQL的Web应用程序,非常容易受到攻击。它的主要目标是成为安全专业人员在合法环境中测试自己的技能和工具的辅助工具,帮助Web开发人员更好地理解保护Web应用程序的过程,并帮助教师/学生在课堂环境中教授/学习Web应用程序安全。DVWA的目标是通过简单直接的界面练习一些最常见的Web漏洞,难度各不相同。请注意,该软件中

