



编辑 | 萝卜皮
蛋白质是身体对抗病原和开发中一种成熟的工具,并用于缩小实验测试的潜在治疗范围。高质量的蛋白质结构是必需的,并且蛋白质经常被视为完全或部分刚性的。
在这里,柏林自由大学(Freie Universität Berlin)的研究人员开发了一个人工智能系统,可以直接从序列信息预测蛋白质-配体复合物的完全柔性全原子结构。
虽然经典对接方法仍然更胜一筹,但这也取决于目标蛋白质的晶体结构。除了预测灵活的全原子结构外,预测置信度指标 (plDDT) 还可用于选择准确的预测,以及区分强结合剂和弱结合剂。
该研究以「Structure prediction of protein-ligand complexes from sequence information with Umol」为题,于 2024 年 5 月 28 日发布在《Nature Communications》。

蛋白质与蛋白质标的接触是评估新药及重新定位已知物的重要问题。现有接触方法存在局限:需要高质量的蛋白质结构;难以确定准确的接触姿态;多基于结合能力(亲和力)评估,难以反映结构稳定性等其他因素。然而,现有的接触方法局限在于需要高质量的蛋白质结构、准确的接触姿态和多基于亲和力评估。因此,采用蛋白质组合和结构评估相结合的方法,限制了对新配体的探索。
机器学习虽然已应用于这一领域,但在针对已知靶标区域的表现上,仍未超越基于打分函数的经典方法。并且,预测的蛋白结构往往不适宜直接用于配体对接。
此外,评估集中若结构基于发布时间而非相似性划分,会引入偏差,尤其是面对训练中未见的受体结构时性能减半。
蛋白质灵活性对于达到结合状态和成功对接至关重要,RoseTTAFold All-Atom 虽能在预测蛋白质时结合配体,其在 PoseBusters 测试集上的成功率也只有 42%,且对未见过的蛋白质表现未知,表明蛋白质-配体复合物结构预测的挑战尚未完全解决。
柏林自由大学的团队开发了一种 AI 方法,通过扩展 AlphaFold2 中的 EvoFormer,可以根据序列信息预测蛋白质-配体复合物的结构。该网络与 RFAA 类似,不同之处在于不包括 3D 轨迹,使用模板结构或额外的晶体学配体数据作为输入或在训练期间使用。
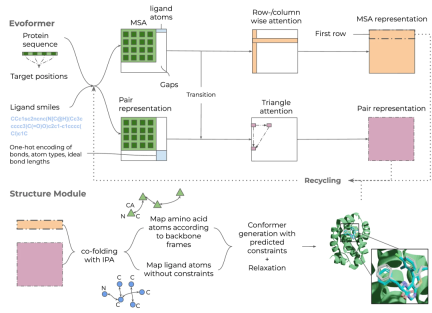
图示:Umol 概述。(来源:论文)
从蛋白质序列、可选蛋白质靶位(口袋)和配体 SMILES 开始,创建了多序列比对 (MSA) 和键矩阵。由此,在网络内生成特征并生成 3D 结构。由于无需任何结构信息即可生成最终的蛋白质-配体复合物结构,因此对蛋白质或配体的灵活性没有任何限制。
与最接近的 RoseTTAFold All-Atom 和 NeuralPlexer1 相比,Umol 在 PoseBusters 测试集上包含口袋信息时获得了更高的成功率(SR,配体 RMSD ≤ 2 Å),分别为 45%、42%、24%,使其成为蛋白质-配体结构预测中表现最好的方法。

图示:预测精度。(来源:论文)
当从 Umol 中删除口袋信息并从 RFAA 中删除模板信息时,SR 分别下降到 18% 和 8%。当使用带有 AF 预测的 DiffDock 时,准确率为 21%,但取决于高度准确的界面预测(口袋 RMSD
许多略高于 2 Å 成功阈值的配体姿势可能相当,这表明可能需要更灵活的评分系统。Umol 在 2.35 Å 阈值下的成功率超过了 AutoDock Vina。在未使用天然蛋白质结构进行评分的情况下,即使是微小的对齐错误也会成为问题。
共折叠蛋白质-配体复合物具有加速药物重新定位的潜力。特别是,研究人员发现配体的预测 lDDT (plDDT) 可用于选择准确的对接姿势,而蛋白质口袋的 pIDDT 适用于选择准确的界面。

图示:置信度指标和准确性。(来源:论文)
配体 plDDT 也分离了高亲和力配体和低亲和力配体,这表明 Umol 和 Umol-pocket 不确定的一些预测可能是弱结合剂。这进一步证明了 Umol 的能力,并强调似乎已经了解了蛋白质-配体相互作用的重要方面。

图示:BindingDB 预测。(来源:论文)
尽管没有口袋信息的准确率为 18%,但网络仍可以在一定程度上区分强结合剂和弱结合剂。这对于注释未知复合物特别有用,该团队以非常高的置信度(配体 plDDT>85)呈现了 336 种蛋白质-配体结构。需要注意的是,虽然这些结构看似合理且其 L-plDDT 得分很高,但仍需通过实验验证。
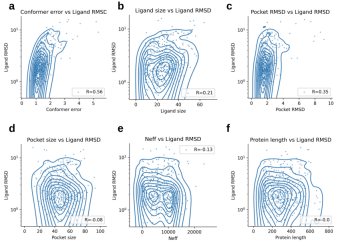
图示:使用 Umol-pocket 分析 PoseBusters 测试集 (n=428) 上的预测的不同特征与配体 RMSD (LRMSD) 之间的关系。(来源:论文)
研究人员没有发现模型的预测性能与「同蛋白质或配体相关的不同特征」之间存在明确的关系。

图示:最困难的 5 个结构。(来源:论文)
然而,在其他方法难以预测的情况下,Umol-pocket 在 5 种情况下有 3 种是准确的。通过反转训练好的网络,可以设计新的配体结合蛋白或蛋白结合配体。另一种选择是使用迁移学习来创建用于相同目的的生成扩散模型。在这种情况下,可以最大化配体或蛋白 plDDT 以尝试创建高亲和力结合物。
PDBbind 的当前版本包含 2019 年从 PDB 处理的数据。从那时起,已经提交了更多蛋白质-配体复合物,这表明可能可以实现更高的精度。
然而,目前尚不清楚需要什么样的精度才能获得有意义的蛋白质-配体对接结果。蛋白质结构预测的高精度在涉及其他分子(如小分子或 RNA)的任务中无法实现。
如果没有蛋白质的共同进化信息,结构预测的准确性会迅速下降。由于小分子或 RNA 没有类似的信息来源,因此人们只能依赖原子表征。
表:PoseBuster 基准集上的成功率(配体 RMSD≤2Å 的百分比)除以 PDBBind 2020 版本的序列同一性 (seqid)。(来源:论文)

研究人员认为口袋信息非常有效,如果没有口袋信息,深度学习方法似乎容易过度拟合。这一发现进一步证实了以下观察结果:尽管 PoseBusters 测试集中的许多分子在训练数据集中包含高度相似的类似物,但这种相似性与模型成功率无关。

图示:一些测试。(来源:论文)
对于基于结构的对接方法(如 Vina 或 Gold),未观察到相同程度的过度拟合。这是意料之中的,因为它们基于原子评分函数,因此不会在相同程度上依赖蛋白质同源性。
深度学习方法在训练集上具有明显更高的性能,这表明蛋白质同源性在蛋白质-配体对接中起着重要作用。RFAA 在测试集上的性能高于训练集,这表明训练集和测试集之间可能存在数据泄漏。
总之,要完全掌握蛋白质-配体相互作用的复杂性还有很长的路要走,但利用深度学习对整个复合物的结构进行预测可能会让科学家更接近解决方案。
Umol:https://github.com/patrickbryant1/Umol
论文链接:https://www.nature.com/articles/s41467-024-48837-6
以上是成功率超越RoseTTAFold系列,用序列信息直接预测蛋白质-配体复合物结构的详细内容。更多信息请关注PHP中文网其他相关文章!

 在LLMS中调用工具Apr 14, 2025 am 11:28 AM
在LLMS中调用工具Apr 14, 2025 am 11:28 AM大型语言模型(LLMS)的流行激增,工具称呼功能极大地扩展了其功能,而不是简单的文本生成。 现在,LLM可以处理复杂的自动化任务,例如Dynamic UI创建和自主a
 多动症游戏,健康工具和AI聊天机器人如何改变全球健康Apr 14, 2025 am 11:27 AM
多动症游戏,健康工具和AI聊天机器人如何改变全球健康Apr 14, 2025 am 11:27 AM视频游戏可以缓解焦虑,建立焦点或支持多动症的孩子吗? 随着医疗保健在全球范围内挑战,尤其是在青年中的挑战,创新者正在转向一种不太可能的工具:视频游戏。现在是世界上最大的娱乐印度河之一
 没有关于AI的投入:获胜者,失败者和机遇Apr 14, 2025 am 11:25 AM
没有关于AI的投入:获胜者,失败者和机遇Apr 14, 2025 am 11:25 AM“历史表明,尽管技术进步推动了经济增长,但它并不能自行确保公平的收入分配或促进包容性人类发展,”乌托德秘书长Rebeca Grynspan在序言中写道。
 通过生成AI学习谈判技巧Apr 14, 2025 am 11:23 AM
通过生成AI学习谈判技巧Apr 14, 2025 am 11:23 AM易于使用,使用生成的AI作为您的谈判导师和陪练伙伴。 让我们来谈谈。 对创新AI突破的这种分析是我正在进行的《福布斯》列的最新覆盖范围的一部分,包括识别和解释
 泰德(Ted)从Openai,Google,Meta透露出庭,与我自己自拍Apr 14, 2025 am 11:22 AM
泰德(Ted)从Openai,Google,Meta透露出庭,与我自己自拍Apr 14, 2025 am 11:22 AM在温哥华举行的TED2025会议昨天在4月11日举行了第36版。它有来自60多个国家 /地区的80个发言人,包括Sam Altman,Eric Schmidt和Palmer Luckey。泰德(Ted)的主题“人类重新构想”是量身定制的
 约瑟夫·斯蒂格利兹(Joseph StiglitzApr 14, 2025 am 11:21 AM
约瑟夫·斯蒂格利兹(Joseph StiglitzApr 14, 2025 am 11:21 AM约瑟夫·斯蒂格利茨(Joseph Stiglitz)是2001年著名的经济学家,是诺贝尔经济奖的获得者。斯蒂格利茨认为,AI可能会使现有的不平等和合并权力恶化,并在几个主导公司的手中加剧,最终破坏了经济的经济。
 什么是图形数据库?Apr 14, 2025 am 11:19 AM
什么是图形数据库?Apr 14, 2025 am 11:19 AM图数据库:通过关系彻底改变数据管理 随着数据的扩展及其特征在各个字段中的发展,图形数据库正在作为管理互连数据的变革解决方案的出现。与传统不同
 LLM路由:策略,技术和Python实施Apr 14, 2025 am 11:14 AM
LLM路由:策略,技术和Python实施Apr 14, 2025 am 11:14 AM大型语言模型(LLM)路由:通过智能任务分配优化性能 LLM的快速发展的景观呈现出各种各样的模型,每个模型都具有独特的优势和劣势。 有些在创意内容gen上表现出色


热AI工具

Undresser.AI Undress
人工智能驱动的应用程序,用于创建逼真的裸体照片

AI Clothes Remover
用于从照片中去除衣服的在线人工智能工具。

Undress AI Tool
免费脱衣服图片

Clothoff.io
AI脱衣机

AI Hentai Generator
免费生成ai无尽的。

热门文章
热工具

SublimeText3汉化版
中文版,非常好用

SublimeText3 Mac版
神级代码编辑软件(SublimeText3)

SecLists
SecLists是最终安全测试人员的伙伴。它是一个包含各种类型列表的集合,这些列表在安全评估过程中经常使用,都在一个地方。SecLists通过方便地提供安全测试人员可能需要的所有列表,帮助提高安全测试的效率和生产力。列表类型包括用户名、密码、URL、模糊测试有效载荷、敏感数据模式、Web shell等等。测试人员只需将此存储库拉到新的测试机上,他就可以访问到所需的每种类型的列表。

Dreamweaver Mac版
视觉化网页开发工具

PhpStorm Mac 版本
最新(2018.2.1 )专业的PHP集成开发工具

