



今年、DeepMind は約 2 億 2,000 万個のタンパク質の予測構造を発表しました。これは、DNA データベース内の既知の生物のほぼすべてのタンパク質をカバーしています。現在、別の巨大テクノロジー企業であるメタ社が、微生物の穴を埋めようとしています。
簡単に言うと、Meta は AI テクノロジーを使用して、細菌やその他のまだ特徴付けられていない微生物の約 6 億個のタンパク質の構造を予測します。チームリーダーのAlexander Rives氏は、「これらのタンパク質は、私たちが最もよく知らない構造であり、非常に謎に満ちたタンパク質です。これらの発見は、生物学をより深く理解する可能性を提供すると思います。」と述べました。 通常、言語モデルは大量のテキストでトレーニングされます。メタ 言語モデルをタンパク質に適用するために、Rives らは、異なる文字で表される 20 個のアミノ酸で構成される既知のタンパク質配列を入力として取り込みました。その後、ネットワークは特定の割合のアミノ酸をマスクしながらタンパク質を自動的に完成させることを学習しました。
メタはこのネットワークに ESMFold という名前を付けました。 ESMFold の予測精度は AlphaFold ほど良くありませんが、構造の予測では AlphaFold より約 60 倍高速です。この速度は、タンパク質構造予測をより大きなデータベースにスケールアップできることを意味します。

- プロジェクトアドレス: https://github.com/facebookresearch/esm
- さて、テストとして、Meta は、土壌、海水、人間の腸、皮膚、その他の微生物の生息地などの環境に由来するメタゲノム DNA のデータベースにモデルを適用することにしました。 Meta AI
また、これは高解像度の予測構造を含む最大のデータベースであり、既存のタンパク質構造データベースの 3 倍の大きさであり、メタゲノムタンパク質を包括的かつ大規模にカバーする最初のデータベースです。
Meta チームは、わずか 2 週間で合計 6 億 1,700 万以上のタンパク質構造を予測しました。 Rives 氏は、予測はモデルの基礎となるコードと同様に無料で誰でも利用できると述べました。 
インタラクティブ バージョンのアドレス: https://esmatlas.com/explore?at=1,1,21.999999344348925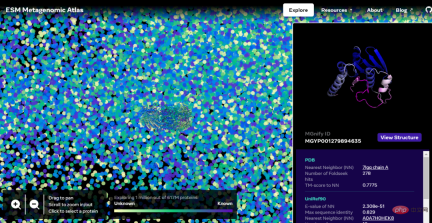
たとえば、下の図は ESMFold による PET 酵素の予測を示しています。
はじめに
ご存知のとおり、タンパク質は遺伝子によってコード化された複雑で動的な分子であり、主に基本的なプロセスを担っています。人生の。タンパク質は生物学において驚くべき役割を果たしています。たとえば、人間の目の桿体と錐体は光を感知して外界を見ることができ、聴覚と触覚の基礎を形成する分子センサー、光エネルギーを化学エネルギーに変換する植物の複雑な分子、分子などです。微生物を動かす「モーター」、人間の筋肉を動かす「モーター」、プラスチックを分解する酵素、病気から私たちを守る抗体などはすべてタンパク質です。 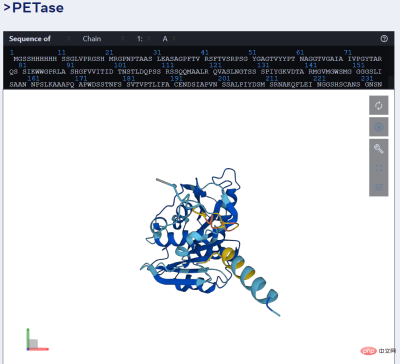
メタゲノミクスにより、科学にとって初めての何十億ものタンパク質配列が明らかになり、NCBI (欧州生物情報学研究所) および共同ゲノム研究所などの公共プロジェクトによって編集された大規模データベースによって初めてカタログ化されました。 。
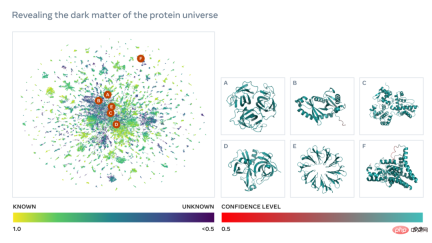
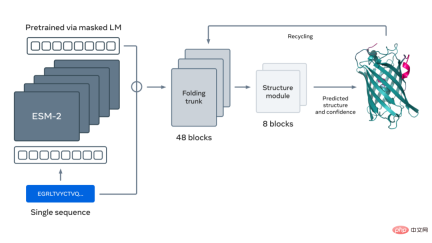
Meta AI によって開発された新しいタンパク質フォールディング手法。大規模言語モデルを活用して、メタゲノム データベース (数億のタンパク質) にタンパク質構造の包括的なビューを初めて作成します。 Meta は、言語モデルが既存の SOTA タンパク質構造予測手法より 60 倍速くタンパク質の原子レベルの三次元構造を予測できることを発見しました。この進歩は、タンパク質構造理解の新時代を加速するのに役立ち、遺伝子配列技術によってカタログ化されている何十億ものタンパク質の構造を理解することが初めて可能になります。 遺伝子配列決定の進歩により、数十億のメタゲノムタンパク質配列の解析が可能になったことはわかっています。カタログ化が可能になります。しかし、数十億のタンパク質の 3D 構造を実験的に決定することは、単一のタンパク質を検出するのに数週間、場合によっては数年かかる X 線結晶構造解析など、時間のかかる実験室技術の範囲をはるかに超えています。コンピューターによるアプローチは、実験的手法では不可能な、メタゲノミクスタンパク質に関する洞察を提供できます。 ESM メタゲノム マップを使用すると、科学者は数億個のタンパク質の規模でメタゲノムタンパク質の構造を検索および分析できるようになります。これは、これまで特徴づけられていなかった構造を特定し、遠い進化の関係を探索し、医学やその他の用途に使用できる新しいタンパク質を発見するのに役立ちます。 以下は、現在知られている構造を持つタンパク質との類似性を示す、数万もの信頼性の高い予測を含むマップです。そして、この画像は初めて、まったく知られていなかったタンパク質構造空間のより広い領域を示しています。 下の図に示すように、ESM-2 言語モデルは予測するようにトレーニングされています。進化の過程 配列によって隠蔽されたアミノ酸。メタ AI は、トレーニングの結果、モデルの内部状態にタンパク質の構造に関する情報が現れることを発見しました。モデルはシーケンスのみでトレーニングされたため、これは本当に驚くべきことです。 論文や手紙のテキストと同じように、タンパク質は一連の文字として書くことができます。各文字は 20 の標準化学元素 (アミノ酸) の 1 つに対応し、それぞれが異なる特性を持ち、タンパク質の構成要素となります。これらの構成要素は天文学的に異なる方法で組み合わせることができます。たとえば、200 個のアミノ酸からなるタンパク質の場合、考えられる配列は 20^200 通りあり、これは目に見える宇宙の原子の数よりも多くなります。各配列は 3D 形状に折り畳まれます (ただし、すべての配列が一貫した構造に折り畳まれるわけではなく、多くは無秩序な形に折り畳まれます)。タンパク質の生物学的機能を大きく決定するのはこの形状です。 生物学的言語を読むことを学ぶことには大きな課題が伴います。タンパク質配列とテキストの一節は両方とも文字として書くことができますが、それらの間には深く根本的な違いがあります。タンパク質配列は、物理法則に従って複雑な 3D 形状に折り畳まれる分子の化学構造を記述します。 タンパク質配列には、タンパク質の折り畳み構造に関する情報を伝える統計的パターンが含まれています。たとえば、タンパク質内の 2 つの位置が共進化する場合、つまり、ある位置で特定のアミノ酸が存在し、通常は他の位置の特定のアミノ酸と対になる場合、これは、その 2 つの位置が同じ位置にあることを意味する可能性があります。折り畳まれた構造の相互作用。これはジグソーパズルの 2 つのピースに似ており、進化では折り畳まれた構造に適合するアミノ酸を選択する必要があります。これは、配列のパターンを観察することでタンパク質の構造を推測できることが多いことを意味します。 ESM は AI を使用してこれらのパターンの読み取りを学習します。 2019 年、メタ AI は、言語モデルがタンパク質の構造や機能などの特性を学習したという証拠を提供しました。マスク言語モデリングと呼ばれる自己教師あり学習の形式を通じて、Meta AI は何百万もの天然タンパク質の配列に基づいて言語モデルをトレーニングしました。この方法を使用すると、モデルはテキスト段落の空白を正しく埋める必要があります (たとえば、「To _ or not to , that is the _____」)。 その後、メタ AI はタンパク質配列のギャップを埋めるために言語モデルをトレーニングしました。彼らは、このトレーニング中にタンパク質の構造と機能に関する情報が得られることを発見しました。 2020年にMetaは、科学者による新型コロナウイルス感染症の進化の予測や病気の遺伝的原因の発見を支援するなど、さまざまな用途向けにSOTAタンパク質言語モデルESM1bをリリースした。 現在、Meta AI はこのアプローチを拡張して、次世代タンパク質言語モデル ESM-2 を作成しました。これは 150 億のパラメータを持ち、これまでで最大のタンパク質言語モデルです。彼らは、モデルのパラメータを 800 万から 150 億にスケールアップすると、内部表現に情報が出現し、原子解像度での 3D 構造予測が可能になることを発見しました。 下の図では、モデルを拡大すると、高解像度のタンパク質構造が表示されます。同時に、モデルが拡大縮小されると、タンパク質構造の原子解像度の画像に新しい詳細が表示されます。 現在の SOTA 計算ツールを使用して、現実的な時間枠で数億のタンパク質配列の構造を予測するには、たとえ大規模な研究を使用したとしても、何年もかかります。組織 リソースについても同様です。したがって、メタゲノムスケールで予測を行うには、予測速度の画期的な進歩が重要です。 Meta AI は、タンパク質配列の言語モデルを使用すると、構造予測が最大 60 倍という大幅な速度で高速化されることを発見しました。これは、わずか数週間でメタゲノム データベース全体の予測を行うのに十分であり、現在公開されているデータベースよりもはるかに大規模なデータベースに拡張することができます。実際、この新しい構造予測機能は、約 2,000 GPU のクラスター上で、わずか 2 週間で 6 億を超えるメタゲノムタンパク質の配列を予測することができました。 さらに、現在の SOTA 構造予測方法では、関連する配列を特定するために大規模なタンパク質データベースを検索する必要があります。これらの方法では、構造関連のパターンを抽出できるように、基本的に進化的に関連したシーケンスのセット全体が入力として必要となります。 Meta AI の ESM-2 言語モデルは、タンパク質配列のトレーニング中にこれらの進化パターンを学習し、タンパク質配列から直接 3D 構造を高解像度で予測できるようにします。 下の図は、ESM-2 言語モデルを使用したタンパク質のフォールディングを示しています。左から右への矢印は、言語モデルから折りたたみトランク、構造モジュールに至るネットワーク内の情報の流れを示し、最後に 3D 座標と信頼度を出力します。 #詳細については、元の記事を参照してください。 ブログリンク: https://ai.facebook.com/blog/protein-folding-esmfold-metagenomics/自然の隠された世界を解き放つ: メタゲノム構造空間の初めての包括的な視点

タンパク質のフォールディングで桁違いの加速を達成

以上がメタ AI が 6 億以上のメタゲノムタンパク質構造マップを開き、150 億の言語モデルが 2 週間で完成の詳細内容です。詳細については、PHP 中国語 Web サイトの他の関連記事を参照してください。

 10生成AIコーディング拡張機能とコードのコードを探る必要がありますApr 13, 2025 am 01:14 AM
10生成AIコーディング拡張機能とコードのコードを探る必要がありますApr 13, 2025 am 01:14 AMねえ、忍者をコーディング!その日はどのようなコーディング関連のタスクを計画していますか?このブログにさらに飛び込む前に、コーディング関連のすべての問題について考えてほしいです。 終わり? - &#8217を見てみましょう
 革新を調理する:人工知能がフードサービスを変革する方法Apr 12, 2025 pm 12:09 PM
革新を調理する:人工知能がフードサービスを変革する方法Apr 12, 2025 pm 12:09 PM食品の準備を強化するAI まだ初期の使用中ですが、AIシステムは食品の準備にますます使用されています。 AI駆動型のロボットは、ハンバーガーの製造、SAの組み立てなど、食品の準備タスクを自動化するためにキッチンで使用されています
 Pythonネームスペースと可変スコープに関する包括的なガイドApr 12, 2025 pm 12:00 PM
Pythonネームスペースと可変スコープに関する包括的なガイドApr 12, 2025 pm 12:00 PM導入 Python関数における変数の名前空間、スコープ、および動作を理解することは、効率的に記述し、ランタイムエラーや例外を回避するために重要です。この記事では、さまざまなASPを掘り下げます
 ビジョン言語モデル(VLM)の包括的なガイドApr 12, 2025 am 11:58 AM
ビジョン言語モデル(VLM)の包括的なガイドApr 12, 2025 am 11:58 AM導入 鮮やかな絵画や彫刻に囲まれたアートギャラリーを歩くことを想像してください。さて、各ピースに質問をして意味のある答えを得ることができたらどうでしょうか?あなたは尋ねるかもしれません、「あなたはどんな話を言っていますか?
 MediaTekは、Kompanio UltraとDimenity 9400でプレミアムラインナップをブーストしますApr 12, 2025 am 11:52 AM
MediaTekは、Kompanio UltraとDimenity 9400でプレミアムラインナップをブーストしますApr 12, 2025 am 11:52 AM製品のケイデンスを継続して、今月MediaTekは、新しいKompanio UltraやDimenity 9400を含む一連の発表を行いました。これらの製品は、スマートフォン用のチップを含むMediaTekのビジネスのより伝統的な部分を埋めます
 今週のAIで:Walmartがファッションのトレンドを設定する前に設定しますApr 12, 2025 am 11:51 AM
今週のAIで:Walmartがファッションのトレンドを設定する前に設定しますApr 12, 2025 am 11:51 AM#1 GoogleはAgent2Agentを起動しました 物語:月曜日の朝です。 AI駆動のリクルーターとして、あなたはより賢く、難しくありません。携帯電話の会社のダッシュボードにログインします。それはあなたに3つの重要な役割が調達され、吟味され、予定されていることを伝えます
 生成AIは精神障害に会いますApr 12, 2025 am 11:50 AM
生成AIは精神障害に会いますApr 12, 2025 am 11:50 AM私はあなたがそうであるに違いないと思います。 私たちは皆、精神障害がさまざまな心理学の用語を混ぜ合わせ、しばしば理解できないか完全に無意味であることが多い、さまざまなおしゃべりで構成されていることを知っているようです。 FOを吐き出すために必要なことはすべてです
 プロトタイプ:科学者は紙をプラスチックに変えますApr 12, 2025 am 11:49 AM
プロトタイプ:科学者は紙をプラスチックに変えますApr 12, 2025 am 11:49 AM今週公開された新しい研究によると、2022年に製造されたプラスチックの9.5%のみがリサイクル材料から作られていました。一方、プラスチックは埋め立て地や生態系に積み上げられ続けています。 しかし、助けが近づいています。エンジンのチーム


ホットAIツール

Undresser.AI Undress
リアルなヌード写真を作成する AI 搭載アプリ

AI Clothes Remover
写真から衣服を削除するオンライン AI ツール。

Undress AI Tool
脱衣画像を無料で

Clothoff.io
AI衣類リムーバー

AI Hentai Generator
AIヘンタイを無料で生成します。

人気の記事
ホットツール

mPDF
mPDF は、UTF-8 でエンコードされた HTML から PDF ファイルを生成できる PHP ライブラリです。オリジナルの作者である Ian Back は、Web サイトから「オンザフライ」で PDF ファイルを出力し、さまざまな言語を処理するために mPDF を作成しました。 HTML2FPDF などのオリジナルのスクリプトよりも遅く、Unicode フォントを使用すると生成されるファイルが大きくなりますが、CSS スタイルなどをサポートし、多くの機能強化が施されています。 RTL (アラビア語とヘブライ語) や CJK (中国語、日本語、韓国語) を含むほぼすべての言語をサポートします。ネストされたブロックレベル要素 (P、DIV など) をサポートします。

SecLists
SecLists は、セキュリティ テスターの究極の相棒です。これは、セキュリティ評価中に頻繁に使用されるさまざまな種類のリストを 1 か所にまとめたものです。 SecLists は、セキュリティ テスターが必要とする可能性のあるすべてのリストを便利に提供することで、セキュリティ テストをより効率的かつ生産的にするのに役立ちます。リストの種類には、ユーザー名、パスワード、URL、ファジング ペイロード、機密データ パターン、Web シェルなどが含まれます。テスターはこのリポジトリを新しいテスト マシンにプルするだけで、必要なあらゆる種類のリストにアクセスできるようになります。

EditPlus 中国語クラック版
サイズが小さく、構文の強調表示、コード プロンプト機能はサポートされていません

SublimeText3 Linux 新バージョン
SublimeText3 Linux 最新バージョン

Dreamweaver Mac版
ビジュアル Web 開発ツール

