

 Technology peripheralsAIEfficiency increased by 1200 times! MIT develops new AI pharmaceutical model
Technology peripheralsAIEfficiency increased by 1200 times! MIT develops new AI pharmaceutical modelEfficiency increased by 1200 times! MIT develops new AI pharmaceutical model

According to foreign media Tech Xplore, researchers at MIT recently developed a new model called EquBind, which can predict the structure of new protein molecules in advance and improve the efficiency of drug development.
At present, this technology has been recognized by the industry, and papers describing this technology will also be accepted by the International Conference on Machine Learning (ICML) in July.
1. The speed is increased by 1200 times, and the EquBind model can quickly screen drug-like molecules
Currently, drug research and development is a long and expensive matter. The main reason is that developing drugs is very expensive. This cost includes not only billions of dollars in capital investment, but also decades of research time.
And during the research and development process, 90% of drugs will fail due to ineffectiveness or too many side effects. Only 10% of drugs can successfully pass the Food and Drug Administration inspection and be approved for marketing.
Therefore, pharmaceutical companies will increase the prices of successfully developed drugs to make up for the losses caused by failed drugs, so the prices of some drugs currently remain high.

▲Some protein molecular structures
If researchers want to develop drugs, they must first find drug-like molecules with development potential. molecules). Another important reason for the slow progress of drug development is the huge number of existing drug-like molecules. Data show that there are currently as many as 1,016 existing drug-like molecules, a number that far exceeds the calculation upper limit of existing molecular calculation models.
In order to process such large molecules with data and speed up the process of drug development, Hannes St rk, a first-year graduate student in the Department of Electrical Engineering and Computer Science at MIT, developed a geometric deep learning model called "EquBind". EquBind runs 1,200 times faster than the fastest existing molecular computational docking models, allowing it to find drug-like molecules faster.
2. The EquBind model can accurately predict protein structure and improve the efficiency of drug development
Currently, most traditional molecular computing docking models use a method called "ligand-protein" (ligand- to-protein binding) method to search for drug-like molecules. Specifically, the model needs to first receive a large number of sample molecules, then let the ligands bind to various molecules, and then the model scores different molecules, and then uses the final ranking to select the most suitable molecules. However, this approach has a complicated process and the model is less efficient in finding drug-like molecules.
Hannes St rk gave a vivid metaphor for this process. He said: "The previous typical 'ligand-protein' approach was like trying to get the model to insert the key into a lock with many keyholes. The model spends a lot of time scoring the fit between the key and each keyhole, and then selecting the most suitable one."
He continued to explain: "And EquBind can skip the most time-consuming steps. , can predict the most suitable 'keyhole' in advance when encountering new molecules, which is called 'blind docking'. EquBind has a built-in geometric reasoning algorithm that can help the model learn the basic structure of the molecule. This algorithm It allows EquBind to directly predict the most suitable position when encountering a new molecule, without spending a lot of time trying different positions and scoring."

▲Massachusetts Polytechnic
3. The EquBind model has been successfully used in the industry, and the author looks forward to more feedback
This model attracted the attention of Pat Walters, chief data officer of the therapeutic company Relay. Notice. Wolster suggested that Hannes Störk's research group use this model to develop drugs for lung cancer, leukemia and gastrointestinal tumors. Generally speaking, protein ligands used in drugs in these fields are difficult to dock using most traditional methods, but EquBind can successfully dock them.

▲Two inhibitor drugs to treat lung cancer
Walters said: "EquBind provides a unique solution to the protein docking problem. It Solving problems such as structure prediction and binding site identification. This method can make good use of thousands of public crystal structure information, and EquBind may impact the field in new ways."
Post this A paper on this technology will be accepted by the International Conference on Machine Learning (ICML) in July. Hannes Strk, the author of the paper, said: "I am looking forward to receiving some suggestions for improving the EquBind model at this conference."
Conclusion: The compatibility between AI and pharmaceuticals is excellent, and the development momentum is booming
AI pharmaceuticals is an emerging field that only entered the public eye in 2020.
The pharmaceutical field is a natural AI scenario. The long cycle, high cost, and low success rate of new drug research and development have left a huge place for AI to be used: machines can learn data independently, mine data, summarize drug research and development rules beyond expert experience, and then optimize the drug research and development process. In all aspects, this can not only improve the efficiency and success rate of drug research and development, but is also expected to reduce research and development expenses and trial and error costs.
Because of such characteristics and development potential, AI pharmaceuticals are currently gaining momentum. However, some people in the industry are pessimistic, saying that AI plays only a supporting role in the pharmaceutical process and cannot circumvent the inherent processes and mechanisms of the industry. It is impossible to complete ten years of work in two or three years.
But overall, there are still new technological breakthroughs in the field of AI pharmaceuticals, and development is booming.
The above is the detailed content of Efficiency increased by 1200 times! MIT develops new AI pharmaceutical model. For more information, please follow other related articles on the PHP Chinese website!

 Are You At Risk Of AI Agency Decay? Take The Test To Find OutApr 21, 2025 am 11:31 AM
Are You At Risk Of AI Agency Decay? Take The Test To Find OutApr 21, 2025 am 11:31 AMThis article explores the growing concern of "AI agency decay"—the gradual decline in our ability to think and decide independently. This is especially crucial for business leaders navigating the increasingly automated world while retainin
 How to Build an AI Agent from Scratch? - Analytics VidhyaApr 21, 2025 am 11:30 AM
How to Build an AI Agent from Scratch? - Analytics VidhyaApr 21, 2025 am 11:30 AMEver wondered how AI agents like Siri and Alexa work? These intelligent systems are becoming more important in our daily lives. This article introduces the ReAct pattern, a method that enhances AI agents by combining reasoning an
 Revisiting The Humanities In The Age Of AIApr 21, 2025 am 11:28 AM
Revisiting The Humanities In The Age Of AIApr 21, 2025 am 11:28 AM"I think AI tools are changing the learning opportunities for college students. We believe in developing students in core courses, but more and more people also want to get a perspective of computational and statistical thinking," said University of Chicago President Paul Alivisatos in an interview with Deloitte Nitin Mittal at the Davos Forum in January. He believes that people will have to become creators and co-creators of AI, which means that learning and other aspects need to adapt to some major changes. Digital intelligence and critical thinking Professor Alexa Joubin of George Washington University described artificial intelligence as a “heuristic tool” in the humanities and explores how it changes
 Understanding LangChain Agent FrameworkApr 21, 2025 am 11:25 AM
Understanding LangChain Agent FrameworkApr 21, 2025 am 11:25 AMLangChain is a powerful toolkit for building sophisticated AI applications. Its agent architecture is particularly noteworthy, allowing developers to create intelligent systems capable of independent reasoning, decision-making, and action. This expl
 What are the Radial Basis Functions Neural Networks?Apr 21, 2025 am 11:13 AM
What are the Radial Basis Functions Neural Networks?Apr 21, 2025 am 11:13 AMRadial Basis Function Neural Networks (RBFNNs): A Comprehensive Guide Radial Basis Function Neural Networks (RBFNNs) are a powerful type of neural network architecture that leverages radial basis functions for activation. Their unique structure make
 The Meshing Of Minds And Machines Has ArrivedApr 21, 2025 am 11:11 AM
The Meshing Of Minds And Machines Has ArrivedApr 21, 2025 am 11:11 AMBrain-computer interfaces (BCIs) directly link the brain to external devices, translating brain impulses into actions without physical movement. This technology utilizes implanted sensors to capture brain signals, converting them into digital comman
 Insights on spaCy, Prodigy and Generative AI from Ines MontaniApr 21, 2025 am 11:01 AM
Insights on spaCy, Prodigy and Generative AI from Ines MontaniApr 21, 2025 am 11:01 AMThis "Leading with Data" episode features Ines Montani, co-founder and CEO of Explosion AI, and co-developer of spaCy and Prodigy. Ines offers expert insights into the evolution of these tools, Explosion's unique business model, and the tr
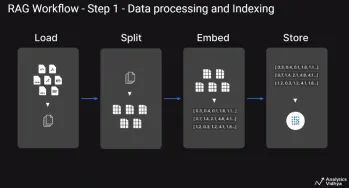 A Guide to Building Agentic RAG Systems with LangGraphApr 21, 2025 am 11:00 AM
A Guide to Building Agentic RAG Systems with LangGraphApr 21, 2025 am 11:00 AMThis article explores Retrieval Augmented Generation (RAG) systems and how AI agents can enhance their capabilities. Traditional RAG systems, while useful for leveraging custom enterprise data, suffer from limitations such as a lack of real-time dat


Hot AI Tools

Undresser.AI Undress
AI-powered app for creating realistic nude photos

AI Clothes Remover
Online AI tool for removing clothes from photos.

Undress AI Tool
Undress images for free

Clothoff.io
AI clothes remover

Video Face Swap
Swap faces in any video effortlessly with our completely free AI face swap tool!

Hot Article
Hot Tools

SecLists
SecLists is the ultimate security tester's companion. It is a collection of various types of lists that are frequently used during security assessments, all in one place. SecLists helps make security testing more efficient and productive by conveniently providing all the lists a security tester might need. List types include usernames, passwords, URLs, fuzzing payloads, sensitive data patterns, web shells, and more. The tester can simply pull this repository onto a new test machine and he will have access to every type of list he needs.

WebStorm Mac version
Useful JavaScript development tools

Atom editor mac version download
The most popular open source editor

EditPlus Chinese cracked version
Small size, syntax highlighting, does not support code prompt function

DVWA
Damn Vulnerable Web App (DVWA) is a PHP/MySQL web application that is very vulnerable. Its main goals are to be an aid for security professionals to test their skills and tools in a legal environment, to help web developers better understand the process of securing web applications, and to help teachers/students teach/learn in a classroom environment Web application security. The goal of DVWA is to practice some of the most common web vulnerabilities through a simple and straightforward interface, with varying degrees of difficulty. Please note that this software

