
Home >Technology peripherals >AI >Chemical retrosynthesis SOTA! Shanghai Jiao Tong University team proposes SMILES alignment technology to achieve efficient retrosynthetic prediction
Chemical retrosynthesis SOTA! Shanghai Jiao Tong University team proposes SMILES alignment technology to achieve efficient retrosynthetic prediction

- 王林Original
- 2024-07-30 20:37:54961browse

Editor | ScienceAI
By using advanced sequence models such as Transformer, the single-step retrosynthesis prediction problem is transformed into a translation task from the SMILES representation of the product to the SMILES representation of the reactant, which has become a widely used strategy with remarkable results.
However, this method often ignores a key point: between the reactants and products, there are a large number of identical substructures that can be directly utilized. Inadequate utilization of these substructures limits the efficiency and accuracy of model predictions.
In July 2024, the research team of Jin Yaohui and Xu Yanyan from the Institute of Artificial Intelligence of Shanghai Jiao Tong University published an article "Ualign: pushing the limit of template-free retrosynthesis prediction with unsupervised SMILES alignment" in the "Journal of Cheminformatics".
In the study, the author proposed a single-step retrosynthetic prediction process, which integrated an unsupervised SMILES sequence alignment technology, aiming to improve the accuracy and efficiency of chemical reaction prediction. The experimental results demonstrate the effectiveness of the model in predicting retrosynthetic pathways and suggest that the model has the potential to become a valuable tool for drug discovery.
Model architecture of Graph to Sequence
If atoms are regarded as nodes, By treating chemical bonds as edges, the molecular structure can be naturally transformed into a graph structure. Compared with sequence models, graph neural networks can better capture the topological structure information inside molecules, thereby achieving more accurate molecular characterization. In addition, compared with other graph structures, chemical bonds in chemical molecules carry rich chemical property information. Based on these advantages, the author proposes a variant based on Graph Attention Network to replace the encoder part in the Transformer model, aiming to provide more powerful molecular representation capabilities for downstream applications.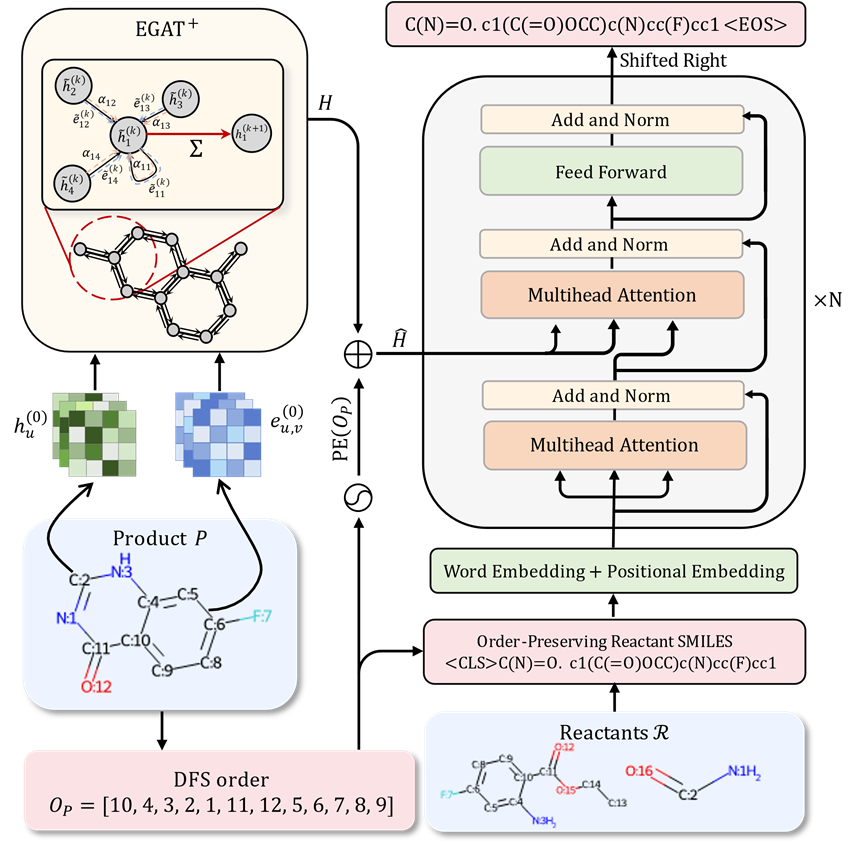
Unsupervised SMILES alignment mechanism
In single-step retrosynthetic prediction, the use of sequence modeling methods usually means that the structure of the reactants must be constructed from scratch, and cannot Make direct modifications based on existing products to efficiently utilize identical substructures between reactants and products. This approach limits the accuracy of generated results to some extent. Considering that the molecular SMILES representation commonly used in sequence modeling actually arranges the atoms and chemical bonds in the molecule in the order of depth-first search, if the position information of each product atom appearing in the reactant SMILES representation can be provided to the model, it will Helps the model identify and reuse substructures that have not changed during the reaction. This will significantly reduce the difficulty for the model to predict reactants and improve the accuracy of predictions. From the perspective of sequence modeling, the commonly used molecular SMILES characterization essentially arranges the atoms and chemical bonds in the molecule according to the order of depth-first search (DFS). If the position information of each atom in the product in the SMILES representation of the reactants can be provided to the model, it will greatly facilitate the model's identification and reuse of unchanged substructures, thereby significantly reducing the difficulty of predicting reactants and improving predictions. accuracy. However, providing this correspondence information directly may introduce the risk of information leakage during model training. To avoid this problem, the researchers proposed an innovative strategy to optimize the model's ability to understand and predict the molecular structure of the reactants without leaking label information. Considering that SMILES sequence characterization is derived from depth-first search on molecular graphs, and most substructures between reactants and products are highly consistent, for a given DFS sequence of any product, there must be a corresponding one The DFS order on the molecular diagram of the reactants is such that the corresponding atoms on the reactants and products appear in almost the same order. Based on this strategy, the researchers not only incorporated the product molecular structure into the model input, but also introduced the DFS order of the reactant molecules as part of the input. In addition, according to the above strategy, the researchers generated a product molecule DFS sequence that is highly consistent with the DFS sequence of a given reactant, and used this sequence to generate a SMILES representation of the reactant as the target of model training. This design allows similar substructures between reactants and products to be arranged in almost the same order in the input and output of the model, thus simplifying the process of the model learning the same structural correspondence between reactants and products, and helping Identify the groups that change during the reaction.Even when the reactant structure is constructed from scratch, this method can effectively reuse product structure information and significantly improve the accuracy of prediction.
Particularly important is that since the DFS order of the product is only based on its molecular structure information and does not rely on any information about the reactants as annotations, this method effectively avoids the problem of label leakage during the model training process.
At the same time, this unsupervised SMILES alignment method does not require the introduction of additional supervision signals during the training process, thereby avoiding complex data annotation and optimization problems in multi-task learning, and provides a novel method for the field of molecular retrosynthesis prediction. and efficient research methods.
Experimental results display
In this study, the author conducted a systematic evaluation of multiple molecular retrosynthesis prediction data sets, covering the widely used USPTO-50K data set, as well as the USPTO-50K data set with a larger amount of data. MIT and USPTO-FULL.
When evaluating model performance, top-k accuracy is used as the main evaluation index. On the USPTO-50K data set, the author not only examined the legality of the SMILES sequence generated by the model, but also conducted a loopback verification of the practical feasibility of the synthesis scheme output by the model through a large-scale pre-trained forward reaction prediction model.
Table 1: Top-k accuracy of USPTO-50K retrosynthetic predictions
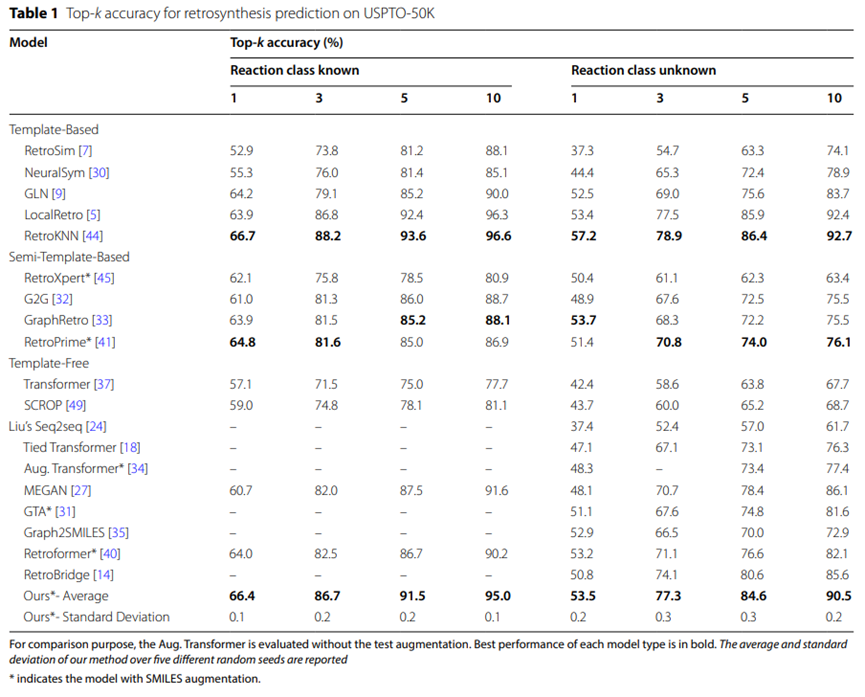
The experimental results of the USPTO-50K data set are summarized in Table 1, showing that the UAlign model performs better in USPTO when the specific reaction type is not specified The top-5 accuracy on the -50K data set is as high as 84.6%, significantly better than other template-free baseline models.
Table 2: Top-k accuracy of USPTO-MIT retrosynthetic prediction
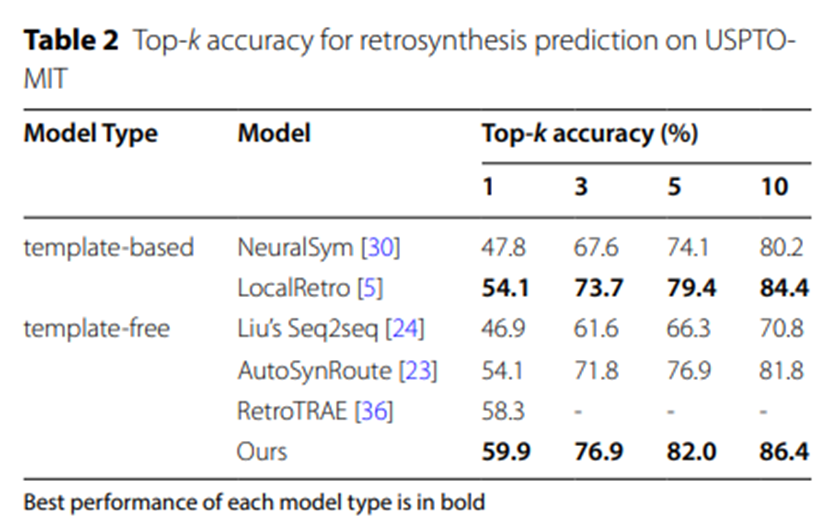
The experimental data in Table 2 and Table 3 further confirm that on the larger-scale data sets USPTO-MIT and USPTO-FULL, UAlign The model surpasses other various baseline models by significant advantages.
Table 3: Top-k accuracy of retrosynthetic prediction on USPTO-FULL
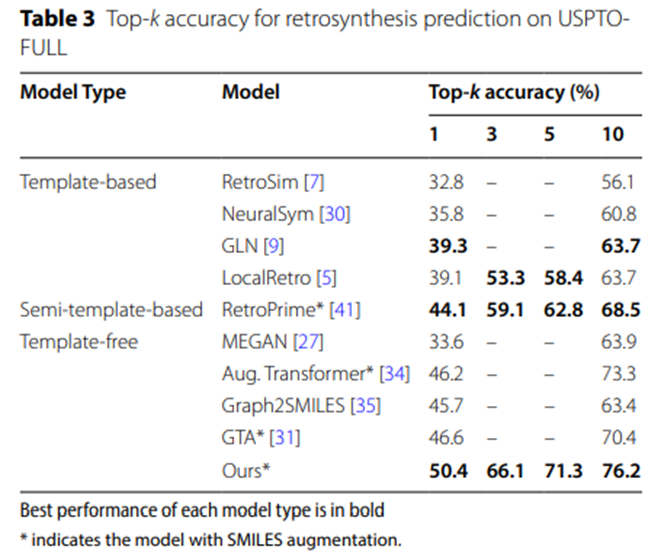
In addition, the experimental results in Table 4 show that compared with other SMILES-based retrosynthetic prediction models, the reactants generated by the UAlign model The SMILES sequence has higher legitimacy.
Table 4: Top-k SMILES effectiveness for retrosynthetic predictions of unknown reaction classes on USPTO-50K
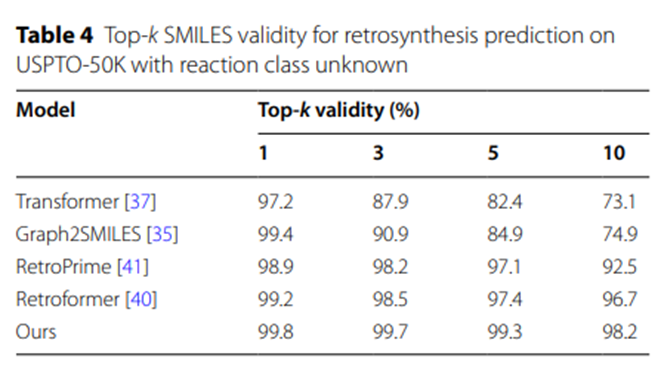
The experimental data in Table 5 further highlights the UAlign model’s ability to generate reasonable and feasible synthesis schemes. Advantage. The reason is that a relatively high proportion of the synthetic schemes proposed by UAlign can pass the verification of the forward reaction prediction model, that is, these schemes can be effectively converted into given target products after corresponding chemical reactions.
Table 5: Top-k round-trip accuracy for retrosynthesis prediction with unknown reaction categories on USPTO-50K
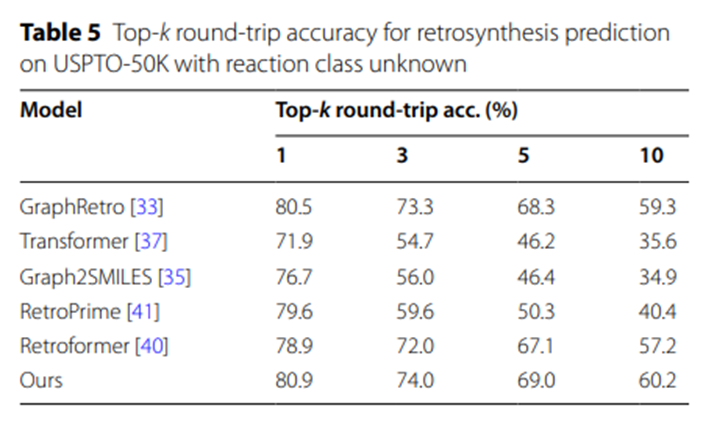
These experimental results not only verify the efficiency and accuracy of the UAlign model in the molecular retrosynthesis prediction task, but also It highlights its excellent performance when processing large-scale data sets and its significant advantages in generating high-quality synthesis solutions.
In order to verify the application potential of the UAlign model in actual production, the author selected new drugs approved by the U.S. Food and Drug Administration (FDA) in the past two years as synthesis targets. Through multiple iterations of the model, the synthesis was successfully obtained. route. The model's predictions of the synthetic routes for these two drugs are highly consistent with the pathways documented in the literature.
In addition, for the third drug, the synthetic route predicted by the model has also been recognized as feasible by experts in the field of chemistry. These synthetic pathways not only cover a variety of reaction types, but also include complex situations such as the synthesis of cyclic compounds and single-step retrosynthetic predictions involving multiple reaction centers.
The above experimental results fully prove that the UAlign model can not only cope with diverse reaction types, but also has high application value in actual production. This shows that the UAlign model has strong practicability and flexibility in the field of molecular retrosynthesis prediction and can provide effective solutions for drug synthesis.

Future outlook
With its excellent performance and flexibility, the UAlign model is fully capable of serving as the cornerstone of building a multi-step retrosynthetic system. It can be combined with various search algorithms and multi-objective optimization technology to form an efficient and intelligent retrosynthetic path planning system.
In addition, the author is also actively exploring the integration of UAlign algorithm with advanced hardware equipment to create an automated unmanned laboratory to promote the automation of drug discovery and synthesis processes, bringing revolutionary changes to the fields of chemical research and drug development. change.
The above is the detailed content of Chemical retrosynthesis SOTA! Shanghai Jiao Tong University team proposes SMILES alignment technology to achieve efficient retrosynthetic prediction. For more information, please follow other related articles on the PHP Chinese website!
Related articles
See more- Technology trends to watch in 2023
- How Artificial Intelligence is Bringing New Everyday Work to Data Center Teams
- Can artificial intelligence or automation solve the problem of low energy efficiency in buildings?
- OpenAI co-founder interviewed by Huang Renxun: GPT-4's reasoning capabilities have not yet reached expectations
- Microsoft's Bing surpasses Google in search traffic thanks to OpenAI technology