
Heim >Technologie-Peripheriegeräte >KI >Neues SOTA für die Vorhersage von Proteinfunktionen, statistikbasierte KI-Methoden vom Shanghai Institute of Technology, Oxford und anderen, veröffentlicht im Nature-Unterjournal
Neues SOTA für die Vorhersage von Proteinfunktionen, statistikbasierte KI-Methoden vom Shanghai Institute of Technology, Oxford und anderen, veröffentlicht im Nature-Unterjournal

- PHPzOriginal
- 2024-08-22 16:45:02826Durchsuche

Proteine verbinden sich mit anderen Molekülen, um nahezu alle grundlegenden biologischen Aktivitäten zu ermöglichen. Daher ist das Verständnis der Proteinfunktion von entscheidender Bedeutung für das Verständnis von Gesundheit, Krankheit, Evolution und Organismusfunktion auf molekularer Ebene.
Allerdings sind mehr als 200 Millionen Proteine noch nicht charakterisiert, und Computermethoden stützen sich in hohem Maße auf Strukturinformationen von Proteinen, um Annotationen unterschiedlicher Qualität vorherzusagen.
Kürzlich hat ein Forschungsteam der Universität Oxford, der ETH Zürich, der Universität Shanghai für Wissenschaft und Technologie und der Beijing Normal University eine statistikbasierte Graphnetzwerkmethode namens PhiGnet entwickelt, um die funktionelle Annotation und die Identifizierung funktioneller Stellen von Proteinen zu fördern.
PhiGnet übertrifft nicht nur andere Methoden in der Leistung, sondern verringert auch die Lücke zwischen Sequenz und Funktion, selbst wenn keine Strukturinformationen vorliegen. Die Ergebnisse zeigen, dass die Anwendung von Deep Learning auf Evolutionsdaten funktionelle Stellen auf der Restebene hervorheben kann und wertvolle Unterstützung für die Interpretation und Untersuchung bestehender Eigenschaften und neuer Funktionen von Proteinen in der Biomedizin bietet.
Relevante Forschung trägt den Titel „Genaue Vorhersage der Proteinfunktion mithilfe statistisch informierter Graphnetzwerke“ und wurde am 4. August in „Nature Communications“ veröffentlicht.

Das Verständnis der Proteinfunktion ist entscheidend für das Verständnis der komplexen Mechanismen vieler wichtiger biologischer Aktivitäten und wichtig für Medizin, Biotechnologie und Pharmazeutika Der Entwicklungsbereich hat weitreichende Auswirkungen.
Bis heute wurden über 356 Millionen Proteine in der UniProt-Datenbank sequenziert (6/2023), von denen die überwiegende Mehrheit (~80 %) keine bekannte funktionelle Annotation aufweist.
Deep-Learning-Methoden haben eine bemerkenswerte Genauigkeit bei der Vorhersage von Protein-3D-Strukturen erreicht und übertreffen die Fähigkeiten klassischer Methoden wie Ab-initio-Methoden und Homologiemodellierung. Allerdings bleibt die genaue Zuordnung funktioneller Anmerkungen zu Proteinen eine Herausforderung, insbesondere im Vergleich zu experimentellen Assays.
Um diese Herausforderungen anzugehen, stellten Forscher die Hypothese auf, dass die in koevolvierenden Resten enthaltenen Informationen zur Annotation von Funktionen auf Restebene verwendet werden könnten.
Das Team der Universität Oxford schlägt vor, statistikbasierte Graphnetzwerke zu verwenden, um Proteinfunktionen nur aus ihren Sequenzen vorherzusagen. Diese Methode charakterisiert von Natur aus evolutionäre Merkmale und ermöglicht eine quantitative Bewertung der Bedeutung von Resten, die bestimmte Funktionen erfüllen.
Diese Methode nutzt Erkenntnisse aus Evolutionsdaten, um zwei Faltungsnetzwerke mit gestapelten Graphen anzutreiben. Mit den gewonnenen Erkenntnissen und der entworfenen Netzwerkarchitektur können Proteinen genaue funktionelle Annotationen zugewiesen werden und, was noch wichtiger ist, die Bedeutung jedes Rests im Verhältnis zu einer bestimmten Funktion quantifiziert werden.
PhiGnet für die funktionelle Annotation von Proteinen
Die PhiGnet-Methode verwendet statistikbasierte Graphennetzwerke, um Proteinfunktionen zu annotieren und funktionelle Stellen artenübergreifend anhand ihrer Sequenzen zu identifizieren.

Um Wissen aus der evolutionären Kopplung (EVC) und der Residuengemeinschaft (RC) zu absorbieren, haben Forscher einen Zweikanal-Architekturansatz unter Verwendung von gestapelten Graph-Faltungsnetzwerken (GCN) entworfen. Diese Methode wurde speziell entwickelt, um Proteinen funktionale Annotationen zuzuweisen, einschließlich der Nummern des Enzymkomitees (EC) und der Begriffe der Genontologie (GO) (biologischer Prozess, BP, zelluläre Komponente, CC und molekulare Funktion, MF).
Wenn eine Proteinsequenz bereitgestellt wird, leitet die Studie ihre Einbettung mithilfe des vorab trainierten ESM-1b-Modells ab. Anschließend werden die Einbettungen als Graphknoten sowie EVCs und RCs (Graphkanten) in sechs Graphfaltungsschichten eines Dual-Stack-GCN eingegeben. Diese Schichten arbeiten mit zwei vollständig verbundenen (FC) Schichtblöcken zusammen, um Informationen von beiden GCNs sorgfältig zu verarbeiten und letztendlich einen Wahrscheinlichkeitstensor zu erzeugen, der die Machbarkeit der Zuweisung funktionaler Annotationen zu Proteinen bewertet.
Darüber hinaus wird der mithilfe der Grad-CAM-Methode (Gradientengewichtete Klassenaktivierungskarte) abgeleitete Aktivierungswert verwendet, um die Bedeutung jedes Rests in einer bestimmten Funktion zu bewerten. Dieser Score ermöglicht es PhiGnet, funktionelle Stellen auf der Ebene der einzelnen Reste zu lokalisieren.
Zum Beispiel wurde durch die Berechnung des RC von Protein D (SdrD), das Serin-Aspartat-Wiederholungen enthält, gezeigt, dass die Reste funktioneller Stellen durch natürliche Evolution erhalten bleiben, und PhiGnet ist in der Lage, solche Informationen zu erfassen und dadurch die Analyse zu verbessern Methoden zur Vorhersage der Proteinfunktion auf Basisebene, auch wenn keine Strukturdaten vorliegen.
Annotieren Sie Proteinfunktionsstellen
Sind rechnerische Vorhersagen genauso genau wie experimentell ermittelte funktionale Anmerkungen? Um diese Frage zu beantworten, verwendete die Studie Aktivierungswerte, um den Beitrag jeder Aminosäure zur Proteinfunktion quantitativ zu untersuchen. Die Vorhersageleistung von PhiGnet wurde bewertet und die Bedeutung von Resten (ihr Beitrag zur Proteinfunktion) in neun Proteinen bewertet.
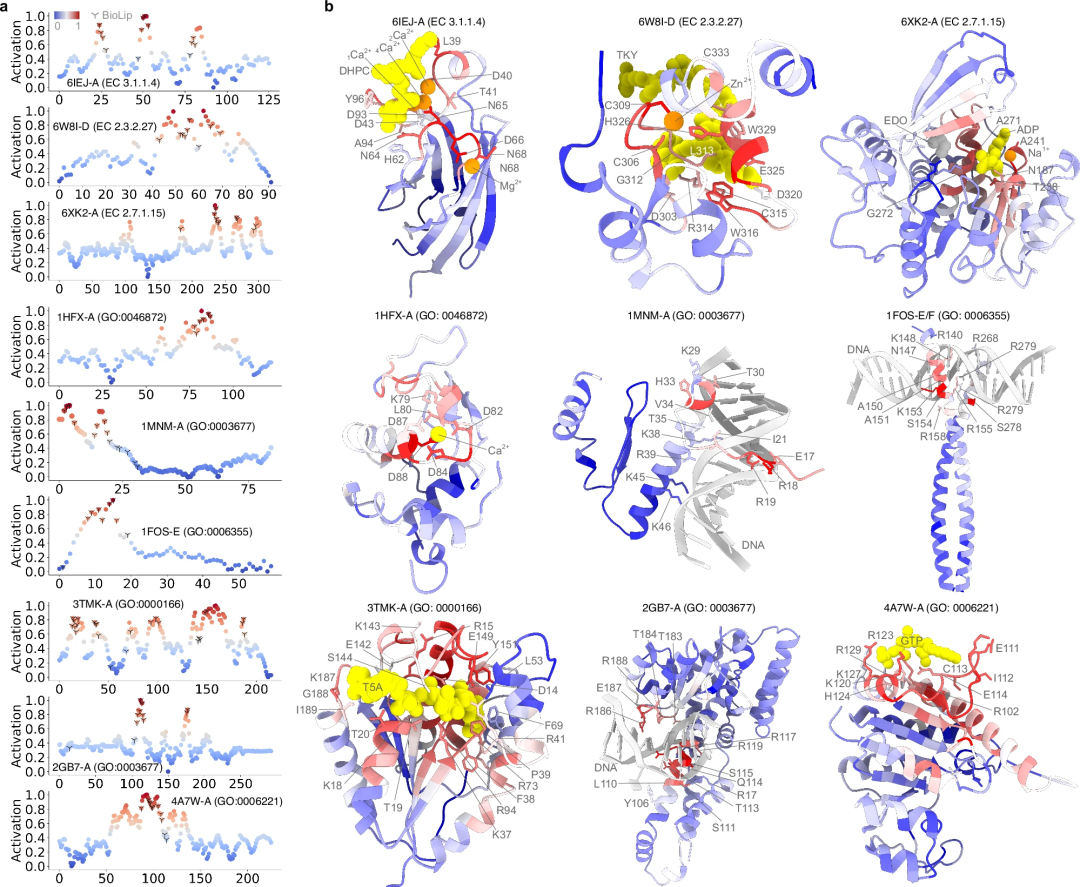
- Durch Berechnung des Aktivierungswerts für jeden Rest in neun Proteinen und Vergleich mit Resten, die entweder experimentell oder halbmanuell ermittelt wurden. Anmerkung. PhiGnet zeigte eine gute Genauigkeit (durchschnittlich ~ 75 %) bei der Vorhersage wichtiger Stellen auf Restebene, in guter Übereinstimmung mit den tatsächlichen Liganden-/Ionen-/DNA-Bindungsstellen. PhiGnet identifiziert funktionell wichtige Reste von Proteinen mit hohen Aktivierungswerten genau.
Übertrifft andere hochmoderne Methoden
- Um die Vorhersageleistung von PhiGnet zu bewerten, wurde die Methode angewendet, um funktionale Annotationen (EC-Zahlen und GO-Terme) von Proteinen in zwei Benchmark-Sets abzuleiten. Vergleichen Sie PhiGnet mit modernsten Methoden, einschließlich ausrichtungsbasierter und Deep-Learning-basierter Methode. Zum Vergleich wurden zwei grundlegende Metriken verwendet, darunter der proteinzentrierte Fmax-Score und die Fläche unter der Precision-Recall-Kurve (AUPR).
Abbildung: Vergleich zwischen verschiedenen Methoden zu GO-Begriffen in verschiedenen Ontologien und EC-Zahlen. (Quelle: Papier)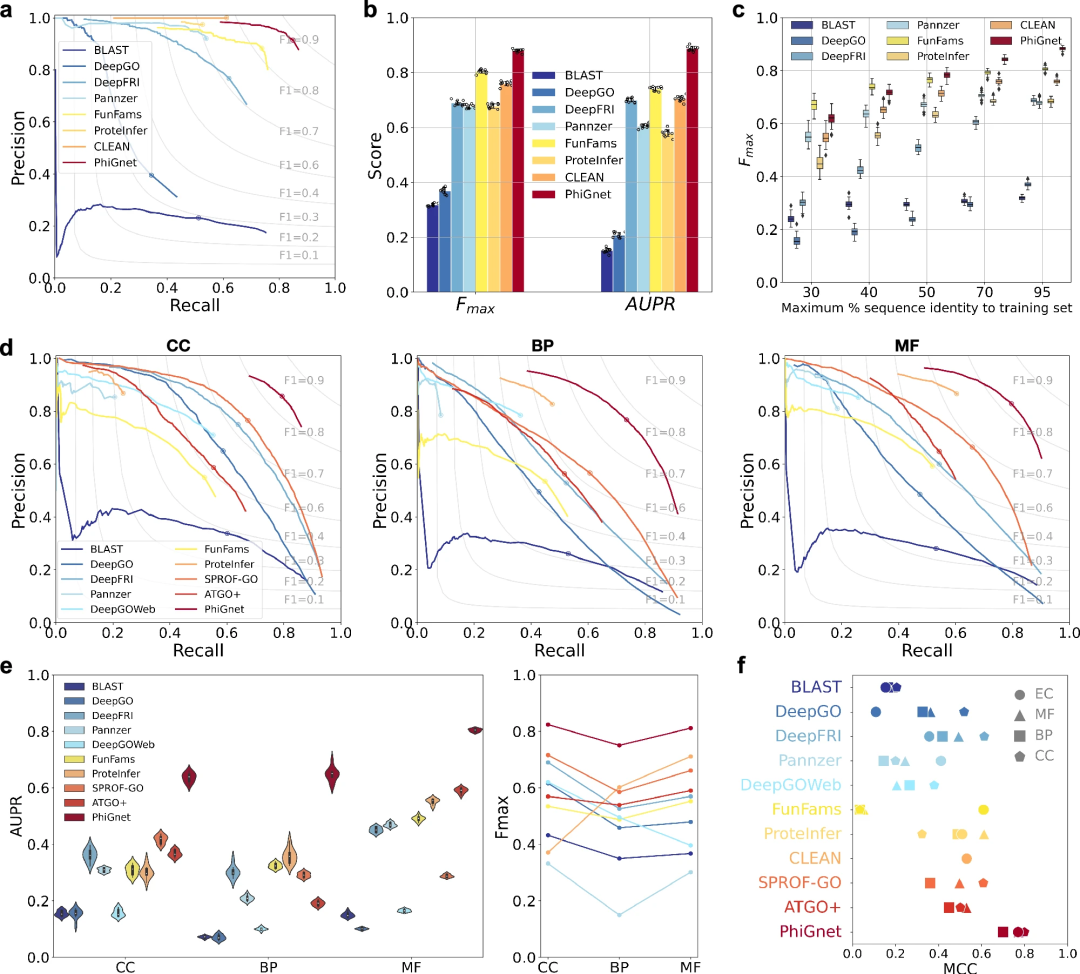
PhiGnet demonstriert die Vorhersagekraft der Zuweisung funktionaler Annotationen zu Proteinen in zwei Testsätzen. Es erreicht einen durchschnittlichen AUPR von 0,70 bzw. 0,89 und Fmax-Werte von 0,80 bzw. 0,88 für GO-Begriffe bzw. EC-Zahlen.
Insgesamt übertrifft PhiGnet alle überwachten und unüberwachten Methoden im Benchmark-Datensatz deutlich.
Darüber hinaus wurde die Generalisierungsrobustheit von PhiGnet beim Testen von Proteinen mit anderen Sequenzidentitätsschwellenwerten als Proteinen im Trainingssatz demonstriert. Bei unterschiedlichen maximalen Sequenzidentitätsniveaus (30 %, 40 %, 50 %, 70 % und 95 %) zeigte PhiGnet mit zunehmender Sequenzidentität eine bessere Vorhersageleistung.
Angetrieben durch evolutionäre Signaturen
Evolutionsdaten spielen eine wichtige Rolle in PhiGnet und können verwendet werden, um funktionelle Annotationen von Proteinen vorherzusagen und funktionelle Stellen zu identifizieren. Zunächst wurden Ablationsexperimente durchgeführt, um den Beitrag von EVC/RC zu PhiGnet zu testen. Experimente zeigen, dass PhiGnet Proteinfunktionsanmerkungen genau zuordnen kann. Darüber hinaus zeigt PhiGnet unter Verwendung von EVC oder RC eine starke Fähigkeit, allgemeine Sequenz-Funktions-Beziehungen zu lernen, oft genauso gut oder genauso gut wie andere Methoden.
Zweitens wurde die Fähigkeit von PhiGnet, sinnvolle Merkmale aus identifizierten funktionsrelevanten Resten in Restgemeinschaften zu charakterisieren, weiter untersucht. Die Aktivierungswerte der Reste wurden berechnet, um ihren Beitrag zur Proteinfunktion hervorzuheben. Bemerkenswerterweise stimmen die vorhergesagten Reste mit denen funktioneller Stellen überein, die durch experimentelle Tests bestimmt wurden, und sind besser identifiziert als diejenigen in RC.
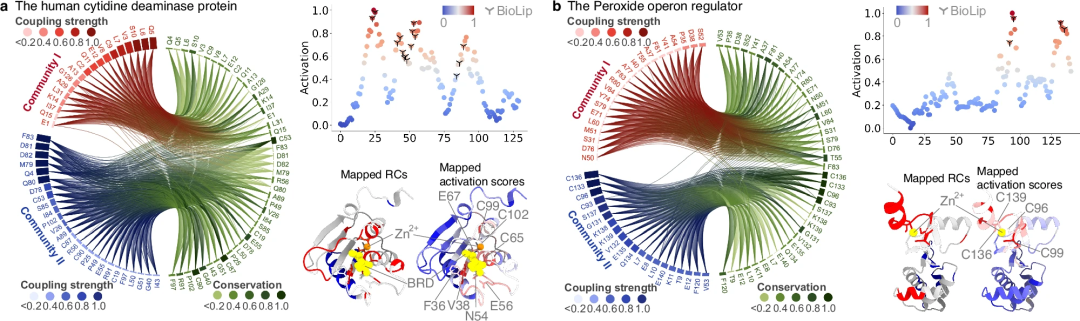
Untersuchungen haben gezeigt, dass evolutionäre Informationen, insbesondere die in der Remote Homology enthaltenen Informationen, ausreichen, um die Funktion eines Proteins zu spezifizieren und die Reste funktioneller Stellen quantitativ zu charakterisieren. Darüber hinaus enthält die Remote Homology höhere Stufen an evolutionärem Wissen im Vergleich zu den niedrigeren Stufen an Informationen im Evolutionary Vector. Gleichzeitig spielen die in Remote Homology enthaltenen Informationen eine wichtige Rolle bei der Verbesserung der Fähigkeit von PhiGnet, funktionsrelevante Stellen auf Restebene zu identifizieren.
Erfolge und Einschränkungen
Zusammenfassend kann die bessere Leistung von PhiGnet auf die Nutzung von Evolutionsdaten von Proteinsequenzen und Mustern höherer Ordnung der Daten zurückgeführt werden, was ein tieferes und genaueres Verständnis der Proteinfunktion ermöglicht. Der Haupterfolg von
PhiGnets Haupterfolg ist die Verwendung statistischer Informationsgraphen-Faltungs-Neuronalnetze, um das hierarchische Lernen von Evolutionsdaten aus riesigen Sequenzdatensätzen zu erleichtern. Dieser Ansatz übertrifft bestehende überwachte und unbeaufsichtigte Methoden erheblich und kann als Leitfaden für zukünftige biologische und klinische Experimente verwendet werden.
Zu den Einschränkungen der PhiGnet-Methode gehört die Verzerrung/das Rauschen, das bei Proteinfamilien mit geringer Sequenzdiversität auftritt. Die Einbeziehung (ko)evolutionärer Informationen in PhiGnet kann sich auf die genaue Identifizierung von Restgemeinschaften auswirken, insbesondere wenn die Informationen aus hochkonservierten Proteinfamilien stammen. Während die Integration physisch extrahierten Wissens in PhiGnet erhebliche Verbesserungen gegenüber anderen Ansätzen erzielt, bleiben erhebliche Herausforderungen bei der Interpretation der Lernmechanismen in PhiGnet bestehen.
Die Synergie zwischen Evolutionsdaten und maschinellem Lernen wird den Weg ebnen, die biophysikalischen Eigenschaften von Proteinen genau zu bestimmen und zu verändern.
Das obige ist der detaillierte Inhalt vonNeues SOTA für die Vorhersage von Proteinfunktionen, statistikbasierte KI-Methoden vom Shanghai Institute of Technology, Oxford und anderen, veröffentlicht im Nature-Unterjournal. Für weitere Informationen folgen Sie bitte anderen verwandten Artikeln auf der PHP chinesischen Website!
In Verbindung stehende Artikel
Mehr sehen- Technologietrends, die Sie im Jahr 2023 im Auge behalten sollten
- Wie künstliche Intelligenz Rechenzentrumsteams neue Alltagsaufgaben beschert
- Können künstliche Intelligenz oder Automatisierung das Problem der geringen Energieeffizienz in Gebäuden lösen?
- OpenAI-Mitbegründer im Interview mit Huang Renxun: Die Argumentationsfähigkeiten von GPT-4 haben noch nicht die Erwartungen erfüllt
- Dank der OpenAI-Technologie übertrifft Bing von Microsoft Google im Suchverkehr