
Heim >Technologie-Peripheriegeräte >KI >SOTA Performance, eine multimodale KI-Methode zur Vorhersage der Protein-Ligand-Affinität in Xiamen, kombiniert erstmals molekulare Oberflächeninformationen
SOTA Performance, eine multimodale KI-Methode zur Vorhersage der Protein-Ligand-Affinität in Xiamen, kombiniert erstmals molekulare Oberflächeninformationen

- WBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOriginal
- 2024-07-17 18:37:101309Durchsuche

Herausgeber |. KX
Im Bereich der Arzneimittelforschung und -entwicklung ist die genaue und effektive Vorhersage der Bindungsaffinität von Proteinen und Liganden für das Arzneimittelscreening und die Arzneimitteloptimierung von entscheidender Bedeutung. Aktuelle Studien berücksichtigen jedoch nicht die wichtige Rolle molekularer Oberflächeninformationen bei Protein-Ligand-Wechselwirkungen.
Auf dieser Grundlage schlugen Forscher der Universität Xiamen ein neuartiges MFE-Framework (Multimodal Feature Extraction) vor, das erstmals Informationen über Proteinoberfläche, 3D-Struktur und -Sequenz kombiniert und einen Kreuzaufmerksamkeitsmechanismus für verschiedene Modi verwendet. Feature-Ausrichtung zwischen Staaten.
Experimentelle Ergebnisse zeigen, dass diese Methode bei der Vorhersage der Protein-Ligand-Bindungsaffinität Spitzenleistungen erbringt. Darüber hinaus belegen Ablationsstudien die Wirksamkeit und Notwendigkeit der Proteinoberflächeninformation und der multimodalen Merkmalsausrichtung innerhalb dieses Rahmens.
Verwandte Forschung mit dem Titel „Oberflächenbasierte multimodale Protein-Ligand-Bindungsaffinitätsvorhersage“ wurde am 21. Juni auf „Bioinformatik“ veröffentlicht.

Forschung zur Vorhersage der Protein-Ligand-Bindungsaffinität
Als Schlüsselphase der Arzneimittelentwicklung wird die Vorhersage der Protein-Ligand-Bindungsaffinität seit langem ausführlich untersucht, was für ein effizientes und genaues Arzneimittelscreening von entscheidender Bedeutung ist.
Herkömmliche computergestützte Tools zur Arzneimittelentdeckung nutzen Scoring-Funktionen (SF), um die Protein-Ligand-Bindungsaffinität grob abzuschätzen, jedoch mit geringer Genauigkeit. Molekulardynamik-Simulationsmethoden können genauere Schätzungen der Bindungsaffinität liefern, sind jedoch oft kostspielig und zeitaufwändig.
Mit der Entwicklung der Computertechnologie und der zunehmenden Fülle umfangreicher biologischer Daten haben Deep-Learning-basierte Methoden großes Potenzial im Bereich der Vorhersage der Protein-Ligand-Bindungsaffinität gezeigt.
Allerdings nutzt die aktuelle Forschung hauptsächlich sequenz- oder strukturbasierte Darstellungen, um die Protein-Ligand-Bindungsaffinität vorherzusagen, und es gibt relativ wenige Studien zu Proteinoberflächeninformationen, die für Protein-Ligand-Wechselwirkungen entscheidend sind.
Eine molekulare Oberfläche ist eine hochrangige Darstellung der Struktur eines Proteins, die charakteristische chemische und geometrische Muster aufweist, die als Fingerabdrücke der Interaktionsmuster des Proteins mit anderen Biomolekülen dienen. Daher begannen einige Studien, Proteinoberflächeninformationen zu nutzen, um die Protein-Ligand-Bindungsaffinität vorherzusagen.
Aber bestehende Methoden konzentrieren sich hauptsächlich auf monomodale Daten und ignorieren die multimodalen Informationen von Proteinen. Darüber hinaus verbinden herkömmliche Methoden bei der Verarbeitung multimodaler Informationen von Proteinen normalerweise Merkmale verschiedener Modalitäten direkt, ohne die Heterogenität zwischen ihnen zu berücksichtigen, was dazu führt, dass die Komplementarität zwischen Modalitäten nicht effektiv genutzt werden kann.
Neuartiges Framework zur multimodalen Merkmalsextraktion
Hier schlagen Forscher ein neuartiges Framework zur multimodalen Merkmalsextraktion (MFE) vor, das zum ersten Mal Informationen aus Proteinoberfläche, 3D-Struktur und Sequenz kombiniert.
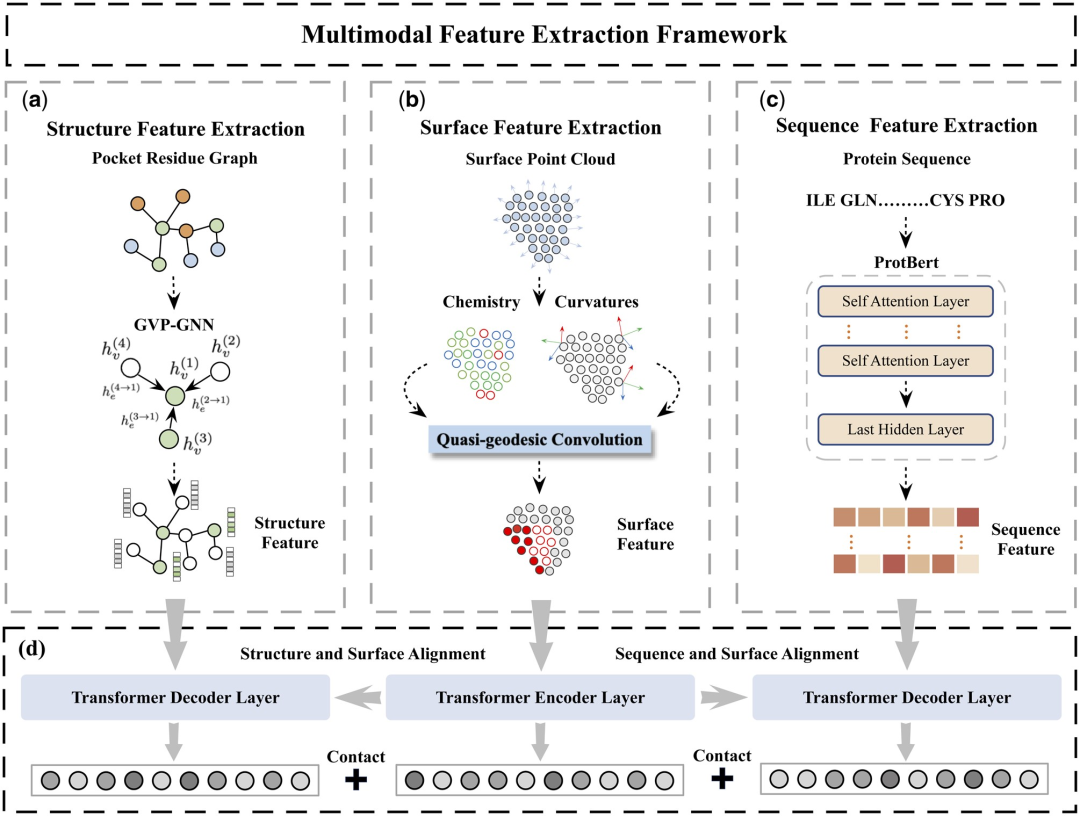
Konkret wurden in der Studie zwei Hauptkomponenten entworfen: das Protein-Merkmalsextraktionsmodul und das multimodale Merkmalsvergleichsmodul.
Das Protein-Feature-Extraktionsmodul wird verwendet, um anfängliche Einbettungen aus Proteinoberflächen-, Struktur- und Sequenzinformationen zu extrahieren.
Im multimodalen Merkmalsvergleichsmodul wird der Kreuzaufmerksamkeitsmechanismus verwendet, um einen Merkmalsvergleich zwischen Proteinstruktur, Sequenzeinbettung und Oberflächeneinbettung zu erreichen und so eine einheitliche und informationsreiche Merkmalseinbettung zu erhalten.
Im Vergleich zu aktuellen Methoden auf dem neuesten Stand der Technik erzielt das vorgeschlagene Framework die besten Ergebnisse bei der Aufgabe der Vorhersage der Protein-Ligand-Bindungsaffinität.
SOTA-Leistung
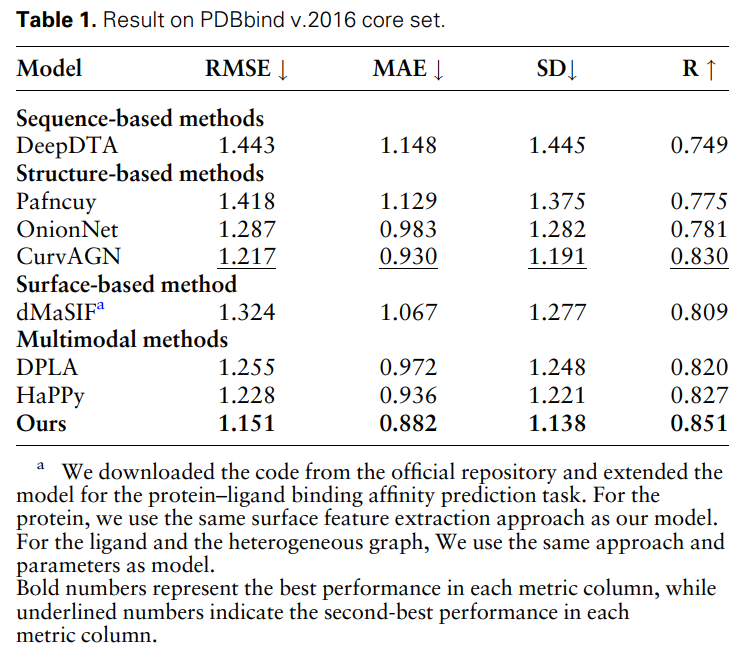
Tabelle 1 zeigt die Ergebnisse von MFE und anderen Basismodellen zur Vorhersage der Protein-Ligand-Bindungsaffinität. Alle Modelle verwendeten die gleiche Partitionierungsmethode für Trainings- und Validierungssätze und wurden auf dem PDBbind-Kernsatz (Version 2016) getestet. Es kann festgestellt werden, dass die MFE-Methode im Vergleich zu allen Basislinien eine SOTA-Leistung erzielt.
Ablationsstudie
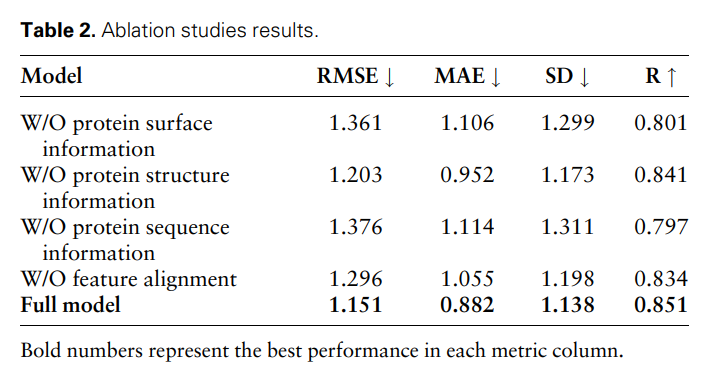
Um die Wirksamkeit und Notwendigkeit verschiedener modaler Merkmale und Merkmalsvergleiche weiter zu beweisen, führten die Forscher die folgenden Ablationsstudien durch: Informationen zur W/O-Proteinoberfläche, Informationen zur W/O-Proteinstruktur, w/ o Proteinsequenzinformationen und merkmalslose Alignments. Die Ergebnisse sind in Tabelle 2 und Abbildung 2 dargestellt.
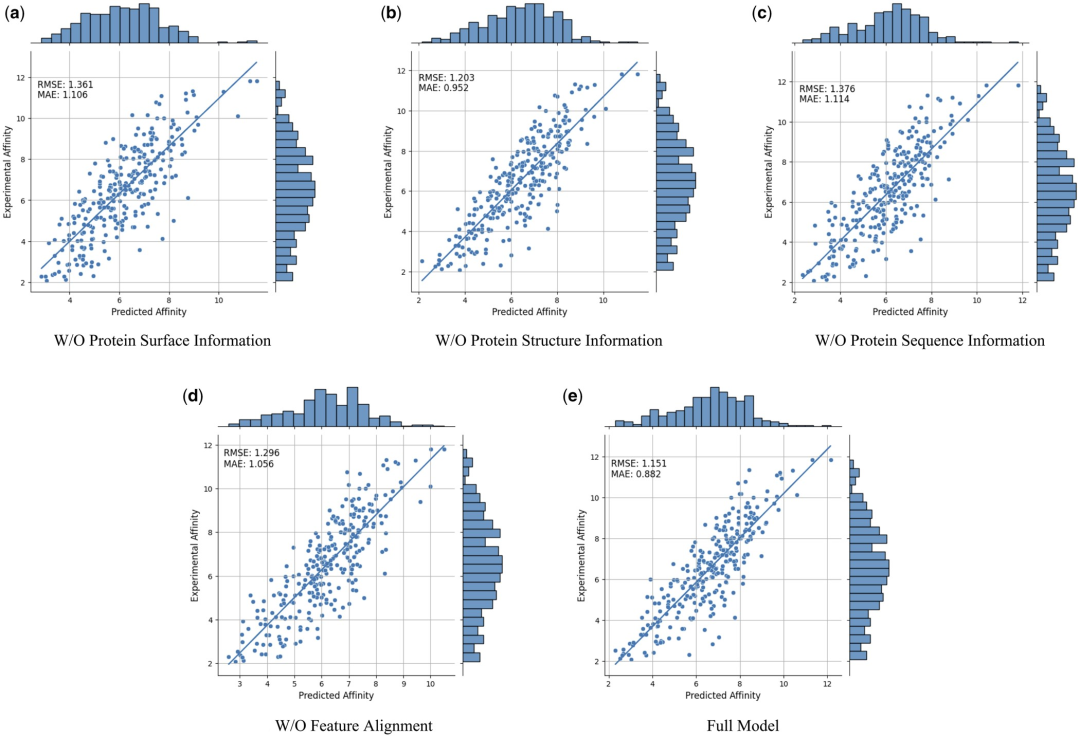
Abbildung 2: Ergebnisse der Ablationsstudie. (Quelle: Papier)
Die Ergebnisse zeigen, dass die Leistung erheblich abnimmt, wenn Oberflächeninformationen entfernt werden, was darauf hindeutet, dass Oberflächeninformationen eine entscheidende Rolle im Modell spielen. Ebenso führt der Ausschluss von Struktur- oder Sequenzinformationen zu einer Leistungsverschlechterung, während die Eliminierung von Sequenzinformationen zu einer stärkeren Verschlechterung führt. Dies liegt daran, dass Sequenzinformationen globale Informationen über das Protein enthalten, die für das vollständige Verständnis des Proteins durch das Modell von entscheidender Bedeutung sind.
Darüber hinaus nimmt ohne Funktionsvergleich die Leistung des Modells ab. Dies unterstreicht die Bedeutung des Merkmalsvergleichs bei der Verarbeitung multimodaler Daten, da er dazu beiträgt, die Heterogenität zwischen verschiedenen Modalmerkmalen zu verringern und dadurch die Fähigkeit des Modells zur effektiven Integration verschiedener Modalmerkmale zu verbessern.
Hyperparameteranalyse
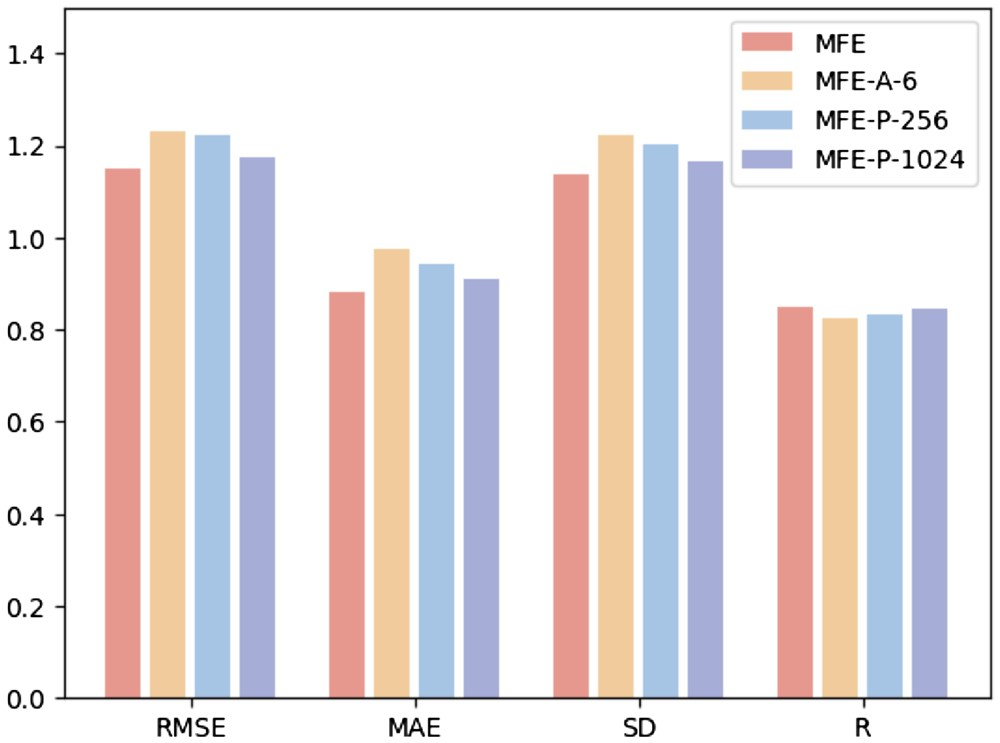
Um den Einfluss verschiedener Hyperparameter auf die Modellleistung zu untersuchen, führten die Forscher die folgenden drei Experimente durch: (i) MFE-A-6: Verwenden Sie nur 6 grundlegende Atomtypen zur Darstellung von Chemikalien Eigenschaften der Oberfläche, einschließlich Wasserstoff, Kohlenstoff, Stickstoff, Sauerstoff, Phosphor und Schwefel; (ii) MFE-P-256: Nur die 256 Oberflächenpunkte, die dem Ligandenzentrum am nächsten liegen, werden als Proteintaschenoberfläche ausgewählt; -P -1024: Wählen Sie die 1024 Oberflächenpunkte, die dem Ligandenzentrum am nächsten liegen, als Proteintaschenoberfläche aus.
Abbildung 3 zeigt die Ergebnisse von drei verschiedenen Hyperparameter-Auswahlmethoden für die Aufgabe zur Vorhersage der Protein-Ligand-Bindungsaffinität.
Merkmalsausrichtungsanalyse und -visualisierung
Um den Einfluss der Merkmalsausrichtung auf die Modellleistung eingehend zu untersuchen, verwendeten die Forscher die Hauptkomponentenanalyse (PCA), um eine Dimensionsreduktion und Summierung der Proteinoberfläche und -struktur durchzuführen und Sequenzmerkmale im Testsatz Visuelle Analyse. Mit diesem Ansatz soll ermittelt werden, ob die Merkmalsausrichtung die Heterogenität zwischen multimodalen Einbettungen verringern kann.
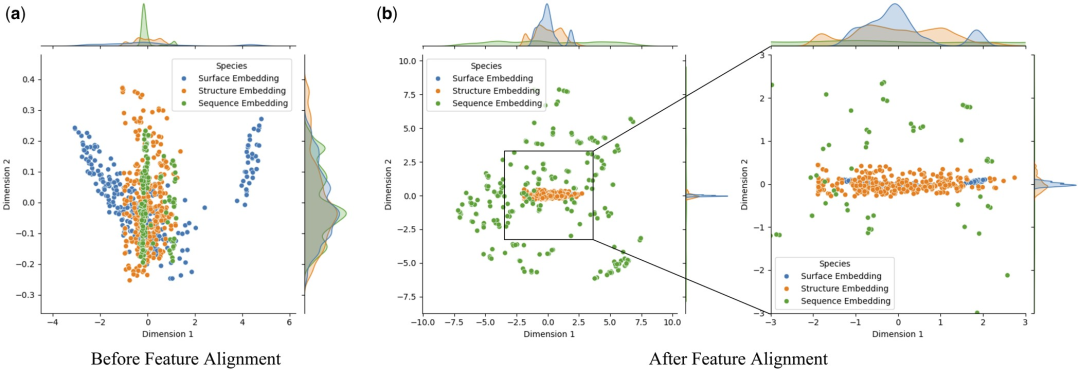
Untersuchungen haben ergeben, dass die Merkmalsausrichtung die Konsistenz zwischen Proteinoberfläche, Struktur und Sequenzeinbettung deutlich verbessert. Dies ist auf die Optimierung multimodaler Feature-Interaktionen in Transformer durch den Aufmerksamkeitsmechanismus zurückzuführen, der Aufmerksamkeitsgewichte zwischen verschiedenen Features berechnet. Dies verbessert die Fähigkeit des Modells, wichtige Informationen zu erfassen, wodurch Daten aus verschiedenen Modalitäten enger im Merkmalsraum geclustert werden können, wodurch Rauschen und Fehler bei der Identifizierung von Protein-Ligand-Wechselwirkungen durch das Modell reduziert werden.
Abschließend kamen die Forscher zu dem Schluss: „Zusammenfassend können wir durch die Untersuchung der Oberfläche von Proteinen ein tieferes Verständnis dafür erlangen, wie Proteine mit anderen Biomolekülen interagieren. In zukünftigen Arbeiten werden wir Proteinoberflächen gründlicher untersuchen, um ihre breitere Anwendung aufzudecken.“ Bioinformatik“
Hinweis: Das Cover stammt aus dem Internet
.Das obige ist der detaillierte Inhalt vonSOTA Performance, eine multimodale KI-Methode zur Vorhersage der Protein-Ligand-Affinität in Xiamen, kombiniert erstmals molekulare Oberflächeninformationen. Für weitere Informationen folgen Sie bitte anderen verwandten Artikeln auf der PHP chinesischen Website!
In Verbindung stehende Artikel
Mehr sehen- Technologietrends, die Sie im Jahr 2023 im Auge behalten sollten
- Wie künstliche Intelligenz Rechenzentrumsteams neue Alltagsaufgaben beschert
- Können künstliche Intelligenz oder Automatisierung das Problem der geringen Energieeffizienz in Gebäuden lösen?
- OpenAI-Mitbegründer im Interview mit Huang Renxun: Die Argumentationsfähigkeiten von GPT-4 haben noch nicht die Erwartungen erfüllt
- Dank der OpenAI-Technologie übertrifft Bing von Microsoft Google im Suchverkehr