



人工智能(AI)一直在迅速发展,但对人类来说,强大的模型却是个「黑匣子」。
我们不了解模型内部的运作原理,不清楚它得出结论的过程。
然而最近,波恩大学(University of Bonn)的化学信息学专家Jürgen Bajorath教授和他的团队取得了重大突破。
他们设计了一种技术,揭示了药物研究中使用的某些人工智能系统的运行机制。
研究显示,人工智能模型主要通过回忆现有数据来预测药物有效性,而非学习特定化学相互作用。
——也就是说,AI预测纯靠拼凑记忆,机器学习实际上并没有学习!
他们的研究结果最近发表在《自然机器智能》(Nature Machine Intelligence)杂志上。

论文地址:https://www.nature.com/articles/s42256-023-00756-9
在医药领域,研究人员正在狂热地寻找有效的活性物质来对抗疾病——哪种药物分子最有效?
通常,这些有效的分子(化合物)会对接在蛋白质上,蛋白质作为触发特定生理作用链的酶或受体。
在特殊情况下,某些分子还负责阻断体内的不良反应,例如过度的炎症反应。
可能的化合物数量巨大,寻找有效的化合物就像大海捞针一样。
因此,研究人员首先使用AI模型来预测,哪些分子最能与各自的靶蛋白对接并牢固结合。然后在实验研究中,更详细地进一步筛选这些候选药物。
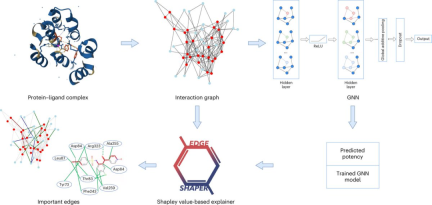
自人工智能发展以来,药物发现研究也越来越多地采用AI相关的技术。
比如图神经网络(GNN),适用于预测某种分子与靶蛋白结合的强度。
图由表示对象的节点和表示节点之间关系的边组成。在蛋白质与配体复合物的图表示中,图的边连接蛋白质或配体节点,表示物质的结构,或者蛋白质和配体之间的相互作用。
GNN模型使用从X射线结构中提取的蛋白质配体相互作用图,来预测配体亲和力。
Jürgen Bajorath教授表示,GNN模型对于我们来说就像一个黑匣子,我们无法得知它如何得出自己的预测。

Jürgen Bajorath教授任职于波恩大学LIMES研究所、波恩-亚琛国际信息技术中心(Bonn-Aachen International Center for Information Technology)和拉玛机器学习与人工智能研究所(Lamarr Institute for Machine Learning and Artificial Intelligence)。
人工智能如何工作?
来自波恩大学化学信息学的研究人员,与罗马Sapienza大学的同事一起,详细分析了图神经网络是否真的学习到了蛋白质与配体的相互作用。
研究人员使用他们专门开发的「EdgeSHAPer」方法分析了总共六种不同的GNN架构。
EdgeSHAPer程序可以判断GNN是否学习了化合物和蛋白质之间最重要的相互作用,或者是通过其他的方式来得出预测。
科学家们使用从蛋白质配体复合物结构中提取的图训练了六个GNN,——化合物的作用方式以及与靶蛋白的结合强度已知。
然后,在其他复合物上测试经过训练的GNN,并使用EdgeSHAPer分析GNN如何产生预测。
「如果GNN按照预期行事,它们需要学习化合物和靶蛋白之间的相互作用,并且通过优先考虑特定的相互作用来给出预测」。
然而,根据研究小组的分析,六个GNN基本上都没有做到这一点。大多数GNN只学会了一些蛋白质与药物的相互作用,主要集中在配体上。
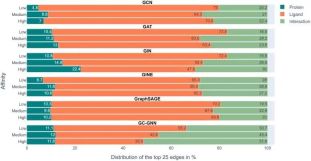
上图展示了在6个GNN中的实验结果,色标条表示用EdgeSHAPer确定的每个预测的前25个边中蛋白质、配体和相互作用所占的平均比例。
我们可以看到,代表绿色的相互作用本该是模型需要学到的,然而在整个实验中所占的比例都不高,而代表配体的橙色条占了最大的比例。
为了预测分子与靶蛋白的结合强度,模型主要「记住」了它们在训练过程中遇到的化学相似分子及其结合数据,而不管靶蛋白如何。这些被记住的化学相似性基本上决定了预测。
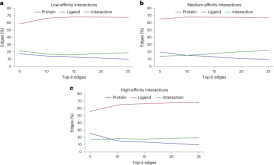
这让人想起「聪明的汉斯效应」(Clever Hans effect),——就像那匹看起来会数数的马,实际上是根据同伴面部表情和手势的细微差别,来推断出预期的结果。
这或许意味着,GNN所谓的「学习能力」可能是站不住脚的,模型的预测在很大程度上被高估了,因为可以使用化学知识和更简单的方法进行同等质量的预测。
不过,研究中也发现了另外一个现象:当测试化合物的效力增加时,模型倾向于学习到更多的相互作用。
也许通过修改表征和训练技术,这些GNN还能朝着理想的方向进一步改进。不过,对于可以根据分子图学习物理量的假设,一般来说应该谨慎对待。
「人工智能不是黑魔法。」
以上是AI未经学习!最新研究揭示了解读人工智能黑盒的方法的详细内容。更多信息请关注PHP中文网其他相关文章!

 让我们跳舞:结构化运动以微调我们的人类神经网Apr 27, 2025 am 11:09 AM
让我们跳舞:结构化运动以微调我们的人类神经网Apr 27, 2025 am 11:09 AM科学家已经广泛研究了人类和更简单的神经网络(如秀丽隐杆线虫中的神经网络),以了解其功能。 但是,出现了一个关键问题:我们如何使自己的神经网络与新颖的AI一起有效地工作
 新的Google泄漏揭示了双子AI的订阅更改Apr 27, 2025 am 11:08 AM
新的Google泄漏揭示了双子AI的订阅更改Apr 27, 2025 am 11:08 AMGoogle的双子座高级:新的订阅层即将到来 目前,访问Gemini Advanced需要$ 19.99/月Google One AI高级计划。 但是,Android Authority报告暗示了即将发生的变化。 最新的Google P中的代码
 数据分析加速度如何求解AI的隐藏瓶颈Apr 27, 2025 am 11:07 AM
数据分析加速度如何求解AI的隐藏瓶颈Apr 27, 2025 am 11:07 AM尽管围绕高级AI功能炒作,但企业AI部署中潜伏的巨大挑战:数据处理瓶颈。首席执行官庆祝AI的进步时,工程师努力应对缓慢的查询时间,管道超载,一个
 Markitdown MCP可以将任何文档转换为Markdowns!Apr 27, 2025 am 09:47 AM
Markitdown MCP可以将任何文档转换为Markdowns!Apr 27, 2025 am 09:47 AM处理文档不再只是在您的AI项目中打开文件,而是将混乱变成清晰度。诸如PDF,PowerPoints和Word之类的文档以各种形状和大小淹没了我们的工作流程。检索结构化
 如何使用Google ADK进行建筑代理? - 分析VidhyaApr 27, 2025 am 09:42 AM
如何使用Google ADK进行建筑代理? - 分析VidhyaApr 27, 2025 am 09:42 AM利用Google的代理开发套件(ADK)的力量创建具有现实世界功能的智能代理!该教程通过使用ADK来构建对话代理,并支持Gemini和GPT等各种语言模型。 w
 在LLM上使用SLM进行有效解决问题-Analytics VidhyaApr 27, 2025 am 09:27 AM
在LLM上使用SLM进行有效解决问题-Analytics VidhyaApr 27, 2025 am 09:27 AM摘要: 小型语言模型 (SLM) 专为效率而设计。在资源匮乏、实时性和隐私敏感的环境中,它们比大型语言模型 (LLM) 更胜一筹。 最适合专注型任务,尤其是在领域特异性、控制性和可解释性比通用知识或创造力更重要的情况下。 SLM 并非 LLMs 的替代品,但在精度、速度和成本效益至关重要时,它们是理想之选。 技术帮助我们用更少的资源取得更多成就。它一直是推动者,而非驱动者。从蒸汽机时代到互联网泡沫时期,技术的威力在于它帮助我们解决问题的程度。人工智能 (AI) 以及最近的生成式 AI 也不例
 如何将Google Gemini模型用于计算机视觉任务? - 分析VidhyaApr 27, 2025 am 09:26 AM
如何将Google Gemini模型用于计算机视觉任务? - 分析VidhyaApr 27, 2025 am 09:26 AM利用Google双子座的力量用于计算机视觉:综合指南 领先的AI聊天机器人Google Gemini扩展了其功能,超越了对话,以涵盖强大的计算机视觉功能。 本指南详细说明了如何利用
 Gemini 2.0 Flash vs O4-Mini:Google可以比OpenAI更好吗?Apr 27, 2025 am 09:20 AM
Gemini 2.0 Flash vs O4-Mini:Google可以比OpenAI更好吗?Apr 27, 2025 am 09:20 AM2025年的AI景观正在充满活力,而Google的Gemini 2.0 Flash和Openai的O4-Mini的到来。 这些尖端的车型分开了几周,具有可比的高级功能和令人印象深刻的基准分数。这个深入的比较


热AI工具

Undresser.AI Undress
人工智能驱动的应用程序,用于创建逼真的裸体照片

AI Clothes Remover
用于从照片中去除衣服的在线人工智能工具。

Undress AI Tool
免费脱衣服图片

Clothoff.io
AI脱衣机

Video Face Swap
使用我们完全免费的人工智能换脸工具轻松在任何视频中换脸!

热门文章
热工具

禅工作室 13.0.1
功能强大的PHP集成开发环境

Atom编辑器mac版下载
最流行的的开源编辑器

Dreamweaver CS6
视觉化网页开发工具

WebStorm Mac版
好用的JavaScript开发工具

DVWA
Damn Vulnerable Web App (DVWA) 是一个PHP/MySQL的Web应用程序,非常容易受到攻击。它的主要目标是成为安全专业人员在合法环境中测试自己的技能和工具的辅助工具,帮助Web开发人员更好地理解保护Web应用程序的过程,并帮助教师/学生在课堂环境中教授/学习Web应用程序安全。DVWA的目标是通过简单直接的界面练习一些最常见的Web漏洞,难度各不相同。请注意,该软件中

