


编辑 | 萝卜皮
蛋白质复合物结构预测在药物研发、抗体设计等应用中发挥着重要作用,然而由于预测精度有限,预测结果与实验结果经常出现不一致。
北京大学、昌平实验室以及哈佛大学的研究团队提出了 ColabDock,这是一个通用框架,它采用深度学习结构预测模型来整合不同形式和来源的实验约束,而无需进一步进行大规模的再训练或微调。
ColabDock 的表现优于使用 AlphaFold2 作为结构预测模型的 HADDOCK 和 ClusPro,不止在具有模拟残基和表面限制的复杂结构预测中,在借助核磁共振化学位移扰动以及共价标记进行的结构预测中也是如此。
另外,它还可以通过模拟界面扫描限制来帮助抗体-抗原界面预测。
该研究以「Integrated structure prediction of protein–protein docking with experimental restraints using ColabDock」为题,于 2024 年 8 月 5 日发布在《Nature Machine Intelligence》。

蛋白质对接为理解生物机制提供了重要的结构信息。尽管深度模型在蛋白质结构预测方面发展迅速,但大多数模型都是以自由对接的方式进行预测,这可能会导致实验约束与预测结构不一致。
为了解决这个问题,北京大学、昌平实验室等机构的研究团队提出了用于受限复合物构象预测的通用框架——ColabDock,它是一个由稀疏实验约束引导的蛋白质-蛋白质对接的通用框架。
通过梯度反向传播,该方法有效地整合了实验约束的先验和数据驱动的蛋白质结构预测模型的能量景观,自动搜索满足两者的构象,同时容忍约束中的冲突或模糊性。
ColabDock 可以利用不同形式和来源的实验约束,而无需进一步进行大规模重新训练或微调。
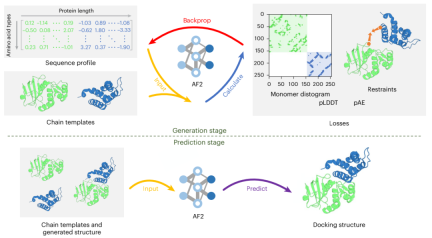
该框架包含两个阶段:生成阶段和预测阶段。
在生成阶段,ColabDock 采用了基于 AlphaFold2 开发的蛋白质设计框架 ColabDesign。在 logit 空间中优化输入序列配置文件,以指导结构预测模型根据给定的实验约束和模板生成复杂结构,同时最大化 pLDDT 和 pAE 测量。
在预测阶段,根据生成的复合物结构和给定的模板预测结构。对于每个目标,ColabDock 会执行多次运行并生成不同的构象。最终构象由排序支持向量机 (SVM) 算法选择。
性能稳健
作为概念验证,研究人员采用 AlphaFold2 作为 ColabDock 中的结构预测模型。当然,这里也可以使用其他数据驱动的深度学习模型,例如 RoseTTAFold2 和 AF-Multimer。
研究人员用合成数据集和几种类型的实验约束上测试 ColabDock,包括 NMR 化学位移扰动 (CSP)、共价标记 (CL) 和模拟深度突变扫描 (DMS)。
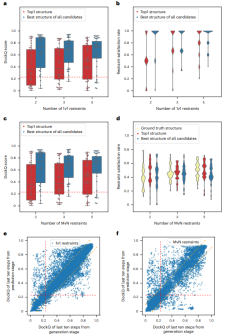
图示:ColabDock 在验证集上的表现。(来源:论文)
ColabDock 评估了两种类型的约束,即 1v1 和 MvN 约束。前者是残基-残基级别的,实例包括来自 XL-MS 的约束。后者是界面级别的,与 NMR 和 CL 实验有关。
在合成数据集上的测试结果表明 ColabDock 取得了令人满意的性能。此外,正如预期的那样,随着约束数量的增加,ColabDock 的性能也得到了提高。
即使只有很少的限制,ColabDock 在基准数据集和相同的框架设置上的表现也优于 AF-Multimer,并且在提供更多限制的情况下收敛到更少的构象,表明有效应用了附加信息。
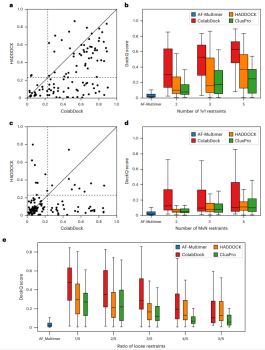
图示:在基准测试集上对 ColabDock、HADDOCK 和 ClusPro 进行比较。(来源:论文)
Compared with HADDOCK and ClusPro, ColabDock performs better when the constraint quality is higher. On both experimental datasets, ColabDock still outperforms HADDOCK and ClusPro regardless of the number and quality of constraints provided.
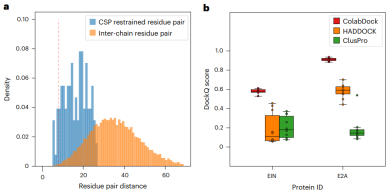
Illustration: Performance and constraint analysis of ColabDock on CSP set. (Source: paper)
Finally, the researchers evaluated the performance of different docking methods on the antibody-antigen data set. ColabDock predicted a much higher proportion of medium or higher quality structures than HADDOCK and ClusPro.
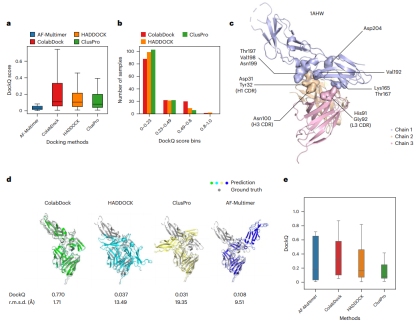
Illustration: Comparison of ColabDock, HADDOCK and ClusPro on the antibody-antigen benchmark set. (Source: paper)
This shows that ColabDock has potential application value in antibody design. Moreover, ColabDock still shows comparable or even better performance than AF-Multimer on the newly released unbiased dataset.
Limitations and Conclusion
ColabDock also has some limitations. Currently, ColabDock can only accept distances smaller than 22 Å, which is determined by the upper limit of the distance map in AlphaFold2. This limitation renders the model applicable to only a small subset of XL-MS reagents.
Without fragment-based optimization, ColabDock can only process complexes of less than 1,200 residues on an NVIDIA A100 graphics processing unit (GPU) due to limited memory.
In addition, this method can be very time-consuming, especially for large protein complexes. Using the bfloat16 floating point format version of AlphaFold2 is expected to help save memory and speed up calculations.
I believe that in the future, after researchers iteratively optimize it, as a unified framework, ColabDock will be able to help bridge the gap between experimental and computational protein science.
Paper link:https://www.nature.com/articles/s42256-024-00873-z
以上是Nature子刊,北大团队通用AI框架对蛋白-蛋白对接进行综合结构预测,弥合实验与计算的差距的详细内容。更多信息请关注PHP中文网其他相关文章!

 让我们跳舞:结构化运动以微调我们的人类神经网Apr 27, 2025 am 11:09 AM
让我们跳舞:结构化运动以微调我们的人类神经网Apr 27, 2025 am 11:09 AM科学家已经广泛研究了人类和更简单的神经网络(如秀丽隐杆线虫中的神经网络),以了解其功能。 但是,出现了一个关键问题:我们如何使自己的神经网络与新颖的AI一起有效地工作
 新的Google泄漏揭示了双子AI的订阅更改Apr 27, 2025 am 11:08 AM
新的Google泄漏揭示了双子AI的订阅更改Apr 27, 2025 am 11:08 AMGoogle的双子座高级:新的订阅层即将到来 目前,访问Gemini Advanced需要$ 19.99/月Google One AI高级计划。 但是,Android Authority报告暗示了即将发生的变化。 最新的Google P中的代码
 数据分析加速度如何求解AI的隐藏瓶颈Apr 27, 2025 am 11:07 AM
数据分析加速度如何求解AI的隐藏瓶颈Apr 27, 2025 am 11:07 AM尽管围绕高级AI功能炒作,但企业AI部署中潜伏的巨大挑战:数据处理瓶颈。首席执行官庆祝AI的进步时,工程师努力应对缓慢的查询时间,管道超载,一个
 Markitdown MCP可以将任何文档转换为Markdowns!Apr 27, 2025 am 09:47 AM
Markitdown MCP可以将任何文档转换为Markdowns!Apr 27, 2025 am 09:47 AM处理文档不再只是在您的AI项目中打开文件,而是将混乱变成清晰度。诸如PDF,PowerPoints和Word之类的文档以各种形状和大小淹没了我们的工作流程。检索结构化
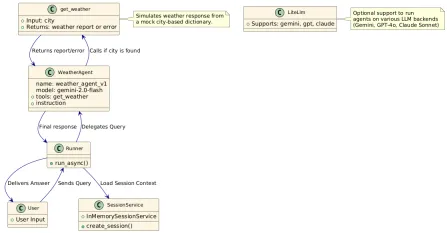 如何使用Google ADK进行建筑代理? - 分析VidhyaApr 27, 2025 am 09:42 AM
如何使用Google ADK进行建筑代理? - 分析VidhyaApr 27, 2025 am 09:42 AM利用Google的代理开发套件(ADK)的力量创建具有现实世界功能的智能代理!该教程通过使用ADK来构建对话代理,并支持Gemini和GPT等各种语言模型。 w
 在LLM上使用SLM进行有效解决问题-Analytics VidhyaApr 27, 2025 am 09:27 AM
在LLM上使用SLM进行有效解决问题-Analytics VidhyaApr 27, 2025 am 09:27 AM摘要: 小型语言模型 (SLM) 专为效率而设计。在资源匮乏、实时性和隐私敏感的环境中,它们比大型语言模型 (LLM) 更胜一筹。 最适合专注型任务,尤其是在领域特异性、控制性和可解释性比通用知识或创造力更重要的情况下。 SLM 并非 LLMs 的替代品,但在精度、速度和成本效益至关重要时,它们是理想之选。 技术帮助我们用更少的资源取得更多成就。它一直是推动者,而非驱动者。从蒸汽机时代到互联网泡沫时期,技术的威力在于它帮助我们解决问题的程度。人工智能 (AI) 以及最近的生成式 AI 也不例
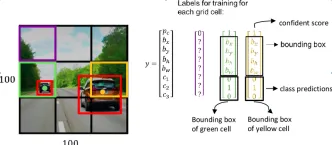 如何将Google Gemini模型用于计算机视觉任务? - 分析VidhyaApr 27, 2025 am 09:26 AM
如何将Google Gemini模型用于计算机视觉任务? - 分析VidhyaApr 27, 2025 am 09:26 AM利用Google双子座的力量用于计算机视觉:综合指南 领先的AI聊天机器人Google Gemini扩展了其功能,超越了对话,以涵盖强大的计算机视觉功能。 本指南详细说明了如何利用
 Gemini 2.0 Flash vs O4-Mini:Google可以比OpenAI更好吗?Apr 27, 2025 am 09:20 AM
Gemini 2.0 Flash vs O4-Mini:Google可以比OpenAI更好吗?Apr 27, 2025 am 09:20 AM2025年的AI景观正在充满活力,而Google的Gemini 2.0 Flash和Openai的O4-Mini的到来。 这些尖端的车型分开了几周,具有可比的高级功能和令人印象深刻的基准分数。这个深入的比较


热AI工具

Undresser.AI Undress
人工智能驱动的应用程序,用于创建逼真的裸体照片

AI Clothes Remover
用于从照片中去除衣服的在线人工智能工具。

Undress AI Tool
免费脱衣服图片

Clothoff.io
AI脱衣机

Video Face Swap
使用我们完全免费的人工智能换脸工具轻松在任何视频中换脸!

热门文章
热工具

安全考试浏览器
Safe Exam Browser是一个安全的浏览器环境,用于安全地进行在线考试。该软件将任何计算机变成一个安全的工作站。它控制对任何实用工具的访问,并防止学生使用未经授权的资源。

禅工作室 13.0.1
功能强大的PHP集成开发环境

mPDF
mPDF是一个PHP库,可以从UTF-8编码的HTML生成PDF文件。原作者Ian Back编写mPDF以从他的网站上“即时”输出PDF文件,并处理不同的语言。与原始脚本如HTML2FPDF相比,它的速度较慢,并且在使用Unicode字体时生成的文件较大,但支持CSS样式等,并进行了大量增强。支持几乎所有语言,包括RTL(阿拉伯语和希伯来语)和CJK(中日韩)。支持嵌套的块级元素(如P、DIV),

SublimeText3汉化版
中文版,非常好用

Atom编辑器mac版下载
最流行的的开源编辑器

