
蛋白質功能預測新SOTA,上海理工、牛津等基於統計的AI方法,登Nature子刊

- PHPz原創
- 2024-08-22 16:45:02869瀏覽

蛋白質與其他分子結合,促進幾乎所有的基礎生物活動。因此,了解蛋白質功能對於理解健康、疾病、演化和分子層面的生物體功能至關重要。
然而,超過 2 億種蛋白質仍未被表徵,計算方法在很大程度上依賴蛋白質的結構資訊來預測不同品質的註釋。
近日,來自牛津大學、蘇黎世聯邦理工學院、上海理工大學和北京師範大學組成的研究團隊,設計了一種基於統計的圖網絡方法,稱為PhiGnet,從而促進蛋白質的功能註釋和功能位點的識別。
PhiGnet 不僅在性能上優於其它方法,而且即使在沒有結構資訊的情況下也縮小了序列-功能差距。研究結果表明,將深度學習應用於演化數據可以突出殘基層級的功能位點,為解釋和研究生物醫學中蛋白質的現有特性和新功能提供寶貴支持。
相關研究以「Accurate prediction of protein function using statistics-informed graph networks」為題,於 8 月 4 日發佈在《Nature Communications》上。

了解蛋白質功能對於理解許多關鍵生物活動的複雜機制至關重要,對醫學、生物技術和藥物開發領域具有深遠的影響。
迄今為止,UniProt 資料庫(6/2023)中已有超過 3.56 億種蛋白質被定序,其中絕大多數(~80%)沒有已知的功能註釋。
深度學習方法在預測蛋白質 3D 結構方面取得了顯著的準確性,超越了從頭算方法和同源性建模等經典方法的能力。然而,準確地將功能註釋分配給蛋白質仍然具有挑戰性,尤其是與實驗測定相比。
為了應對這些挑戰,研究人員假設可以利用共同演化殘基中所包含的資訊來註釋殘基層級的功能。
牛津大學團隊提出利用基於統計的圖網絡僅從蛋白質序列預測其功能。此方法固有地表徵了演化特徵,可以對執行特定功能的殘基的重要性進行定量評估。
此方法利用從演化資料中獲得的知識來驅動兩個堆疊圖卷積網路。借助所獲得的知識和設計的網路架構,可以準確地為蛋白質分配功能註釋,並且重要的是,可以量化每個殘基相對於特定功能的重要性。
用於蛋白質功能註釋的 PhiGnet
PhiGnet 方法使用基於統計的圖網絡來註釋蛋白質功能並根據其序列識別跨物種的功能位點。

為了從演化耦合(EVC)和殘基群落(RC)中吸收知識,研究人員設計了雙通道架構的方法,採用堆疊圖卷積網路 (GCN)。此方法專門用於為蛋白質分配功能註釋,包括酵素委員會 (EC) 編號和基因本體 (GO) 術語(生物過程、BP、細胞組成、CC 和分子功能、MF)。
當提供蛋白質序列時,研究使用預先訓練的 ESM-1b 模型得出其嵌入。隨後,將嵌入作為圖節點以及 EVC 和 RC(圖邊)輸入到雙堆疊 GCN 的六個圖卷積層中。這些層與兩個完全連接 (FC) 層塊協同工作,精心處理來自兩個 GCN 的信息,最終生成一個概率張量,用於評估為蛋白質分配功能註釋的可行性。
此外,使用梯度加權類別活化圖 (Grad-CAM) 方法得出的活化分數(activation score)用於評估每個殘基在特定功能中的重要性。此分數使 PhiGnet 能夠在單一殘基層級上精確定位功能位點。
例如,通過計算含有絲氨酸-天冬氨酸重複序列的蛋白質D (SdrD) 的RC,表明功能位點的殘基通過自然進化而得以保留,並且PhiGnet 能夠捕獲此類信息,從而改進在殘基底水平上預測蛋白質功能的方法,即使在沒有結構數據的情況下也是如此。
註釋蛋白質功能位點
計算による予測は、実験的に決定された機能アノテーションと同じくらい正確ですか?この疑問に対処するために、この研究では活性化スコアを使用して、タンパク質の機能に対する各アミノ酸の寄与を定量的に調べました。 PhiGnet の予測パフォーマンスが評価され、9 つのタンパク質における残基の重要性 (タンパク質の機能への寄与) が評価されました。
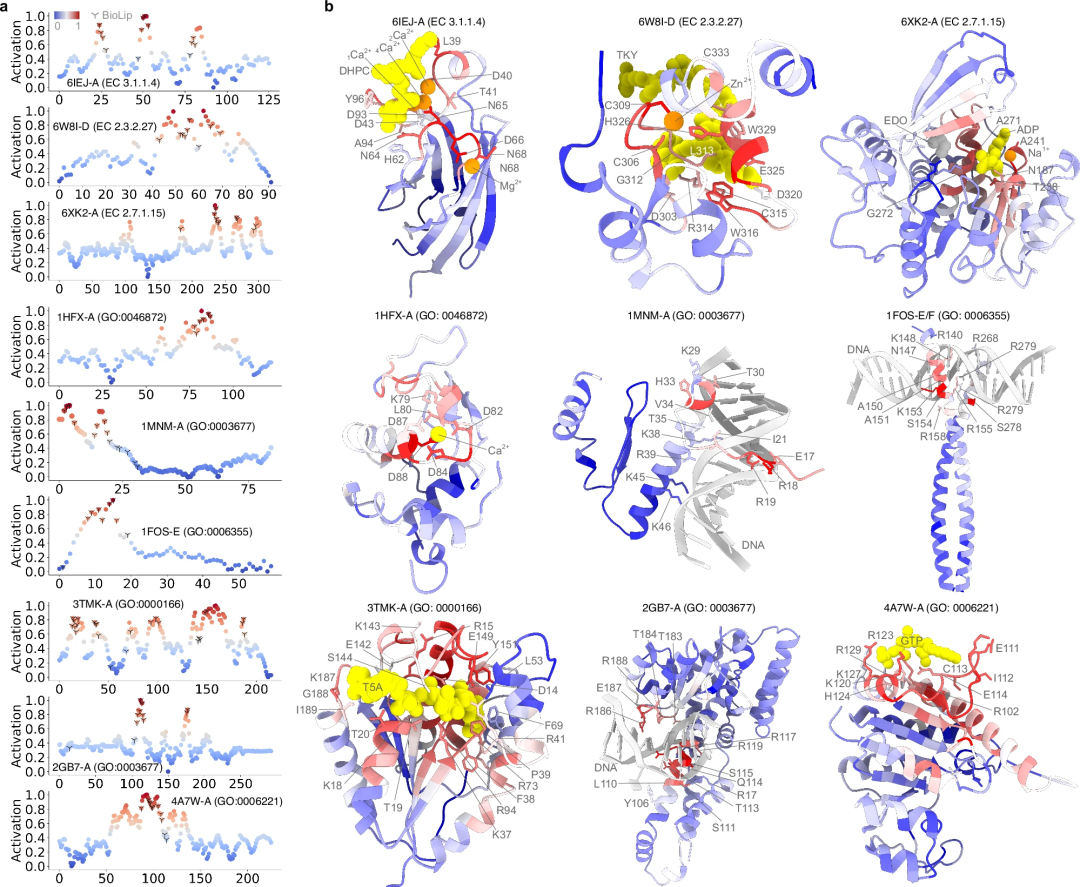
- 9 つのタンパク質の各残基の活性化スコアを計算し、実験的または半手動でアノテーションを付けて決定された残基と比較します。 PhiGnet は、残基レベルで重要な部位を予測する際に良好な精度 (平均 ⩾ 75%) を示し、実際のリガンド/イオン/DNA 結合部位とよく一致しました。 PhiGnet は、高い活性化スコアを持つタンパク質の機能的に重要な残基を正確に識別します。
他の最先端のメソッドを上回るパフォーマンス
- PhiGnet の予測パフォーマンスを評価するために、このメソッドを適用して 2 つのベンチマーク セット内のタンパク質の機能アノテーション (EC 番号と GO ターム) を推論しました。 PhiGnet を、アライメントベースのメソッドやディープラーニングベースのメソッドなどの最先端のメソッドと比較します。比較には、タンパク質中心の Fmax スコアと精度再現率曲線下面積 (AUPR) を含む 2 つの基本指標が使用されました。
図: 異なるオントロジーおよび EC 番号における GO 用語の異なる手法間の比較。 (出典: 論文)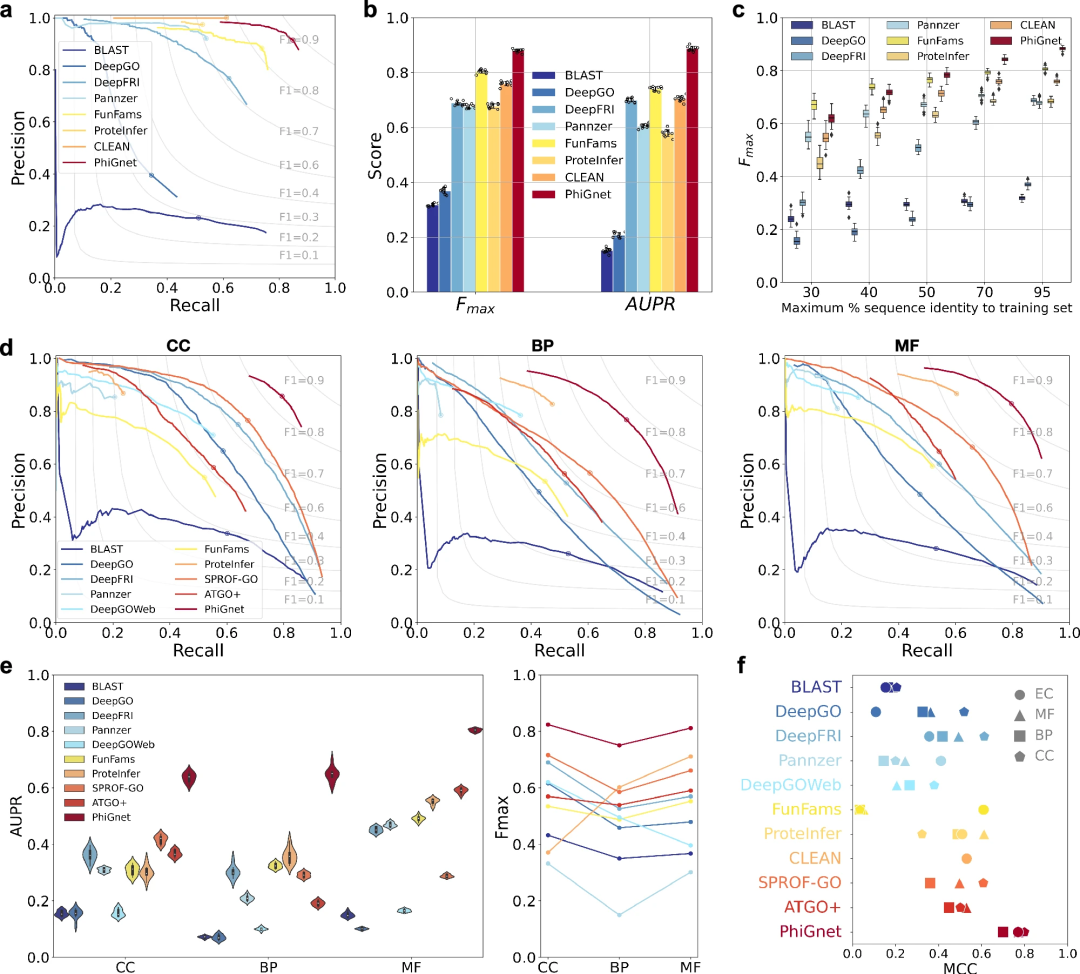
PhiGnet は、2 つのテスト セットでタンパク質に機能的アノテーションを割り当てる予測能力を実証します。 GO タームと EC 番号の平均 AUPR はそれぞれ 0.70 と 0.89、Fmax スコアは 0.80 と 0.88 を達成しています。
全体として、PhiGnet はベンチマーク データセット上ですべての教師ありメソッドと教師なしメソッドを大幅に上回っています。
さらに、PhiGnet の一般化堅牢性は、トレーニング セット内のタンパク質とは異なる配列同一性閾値を持つタンパク質をテストすることが実証されました。異なる最大配列同一性レベル (30%、40%、50%、70%、および 95%) で、PhiGnet は配列同一性が増加するにつれてより優れた予測パフォーマンスを示しました。
進化的特徴による駆動
進化的データは PhiGnet において重要な役割を果たしており、タンパク質の機能的アノテーションを予測し、機能部位を特定するために使用できます。まず、PhiGnet への EVC/RC の貢献をテストするためにアブレーション実験が実行されました。実験により、PhiGnet がタンパク質の機能アノテーションを正確に割り当てることができることが示されました。さらに、EVC または RC を使用する PhiGnet は、多くの場合、他の方法と同様に、一般的なシーケンスと関数の関係を学習する強力な能力を示します。
第二に、残基コミュニティで特定された機能的に関連する残基から意味のある特徴を特徴付ける PhiGnet の能力がさらに調査されました。残基の活性化スコアは、タンパク質機能への寄与を強調するために計算されました。注目すべきことに、予測された残基は実験的アッセイによって決定された機能部位の残基と一致しており、RC のものよりもよく同定されています。
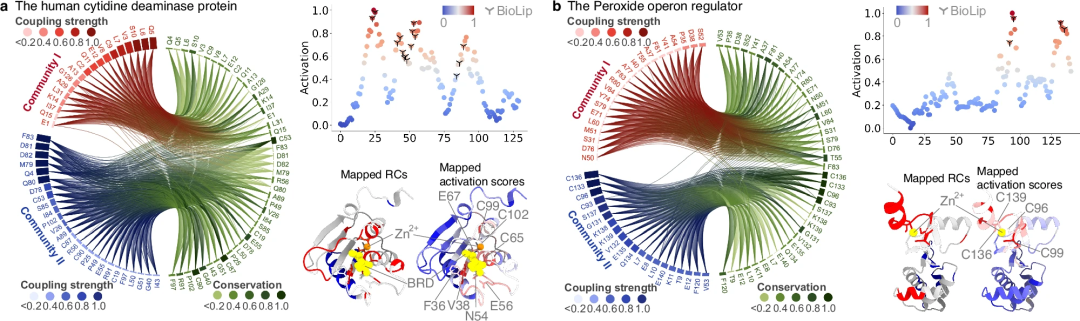
研究により、進化情報、特にリモートホモロジーに含まれる情報は、タンパク質の機能を特定し、機能部位の残基を定量的に特徴付けるのに十分であることが示されています。さらに、リモート相同性には、進化ベクトルの下位レベルの情報と比較して、高次レベルの進化の知識が含まれています。同時に、Remote Homology に含まれる情報は、残基レベルで機能的に関連する部位を特定する PhiGnet の能力を強化する上で重要な役割を果たします。
成功と限界
要約すると、PhiGnet のパフォーマンスが向上したのは、タンパク質配列の進化データとそのデータの高次パターンを利用し、タンパク質の機能をより深く正確に理解できるようになったことに起因すると考えられます。
PhiGnet の主な成功は、統計情報グラフ畳み込みニューラル ネットワークを使用して、大規模なシーケンス データセットからの進化データの階層学習を促進したことです。このアプローチは、既存の教師ありおよび教師なしの方法を大幅に上回り、将来の生物学的実験や臨床実験の指針として使用できます。
PhiGnet メソッドの制限には、配列多様性の低いタンパク質ファミリーで発生するバイアス/ノイズが含まれます。 (共)進化情報を PhiGnet に組み込むと、特にその情報が高度に保存されたタンパク質ファミリーに由来する場合、残基コミュニティの正確な同定に影響を与える可能性があります。物理的に抽出された知識を PhiGnet に統合すると、他のアプローチに比べて大幅な改善が得られますが、PhiGnet の学習メカニズムを解釈する際には大きな課題が残っています。
進化データと機械学習の相乗効果により、タンパク質の生物物理学的特性を正確に決定し、操作する道が開かれます。
以上是蛋白質功能預測新SOTA,上海理工、牛津等基於統計的AI方法,登Nature子刊的詳細內容。更多資訊請關注PHP中文網其他相關文章!