
기하학적 딥러닝 방법을 사용하여 약물 분자 합성을 위한 최적 옵션을 예측하여 신약 발견의 길을 열었습니다.

- WBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWB앞으로
- 2024-01-13 22:36:071126검색

후기능화는 약물 후보물질의 특성을 최적화하는 경제적인 방법입니다. 그러나 약물 분자의 화학적 복잡성으로 인해 종종 후기 단계 기능화가 어려워집니다.
이 문제를 해결하기 위해 뮌헨 대학, ETH Zurich 및 Roche Basel의 연구자들은 협력하여 후기 단계 기능화 플랫폼을 기반으로 합니다. 기하학적 딥 러닝 및 고처리량 반응 스크리닝 기술
보릴화는 기능화의 핵심 단계 중 하나라는 점을 고려하여 계산 모델을 사용하여 평균 절대 오차 범위가 4~5%인 다양한 반응 조건에서 수율을 예측했습니다. 이 모델은 각각 92%와 67%의 정확도로 알려진 기질과 알려지지 않은 기질에 대한 새로운 반응을 분류할 수 있었습니다. 우리는 분류기의 F 점수가 67%로 주요 제품의 위치 선택성을 정확하게 포착할 수 있었습니다. 23개의 다양한 상업용 약물 분자에 적용했을 때 우리는 구조적 다양화를 위한 많은 기회를 성공적으로 발견했습니다
이 연구는 "기하학적 딥러닝을 사용하여 고처리량 실험을 활성화하여 후기 단계 약물 다양화 촉진"이라는 제목으로 2023년 11월에 발표되었습니다. 3월 23일 Nature Chemistry 저널에 게재됨

LSF 프로젝트는 의약 화학 연구에서 중요한 역할을 합니다.
의약 화학에서 구조-활성 관계를 확립하는 것을 목표로 할 때 구조의 참신함과 복잡성으로 인해 화학 표적 구조 합성이 어려워집니다. 구조-활성 관계 모델은 선도 화합물 및 선도 화합물 최적화 계획을 안내하여 약물 후보의 약리학적 활성 및 물리화학적 특성을 개선할 수 있습니다. 구조-활동 관계를 탐색하려면 설계-제작-테스트-분석 주기의 병목 현상인 효율적인 통합이 중요합니다.
유기 지지체의 후기 단계 기능화를 위해 C-H 결합을 활성화하고 수정하는 많은 대체 방법이 있습니다( LSF)는 분자 빌딩 블록부터 고급 제약 분자까지 다양합니다. 많은 촉매 시스템은 변형된 유사체에 대한 화학적 및 위치 선택적 접근뿐만 아니라 방향성 및 비방향성 접근 방식을 제공합니다.
수많은 LSF 방법 중에서 C-H 보릴화 방법은 신속한 화합물 다양화를 위해 가장 일반적으로 사용되는 방법으로 간주됩니다. 유기 붕소 화합물은 후속 C-C 결합 커플링 반응을 위한 신뢰할 수 있는 수단으로 다양한 작용기로 전환될 수 있으므로 광범위한 구조-활성 관계 연구가 가능합니다
그러나 현재 약물 발견 보고서에서 LSF의 적용은 소수에 불과합니다. 이러한 보고서의 대부분은 단일 LSF 반응 유형에 중점을 둡니다. 서로 다른 결합 강도, 전자 특성, 입체 및 작용기 환경을 갖는 여러 유형의 C-H 결합의 직접 LSF는 문제를 야기합니다. 더욱이, LSF 프로젝트를 수행하는 것은 종종 시간과 자원 집약적이며, 이는 많은 의약 화학 프로젝트의 빡빡한 일정과 제한된 자산과 일치하지 않습니다
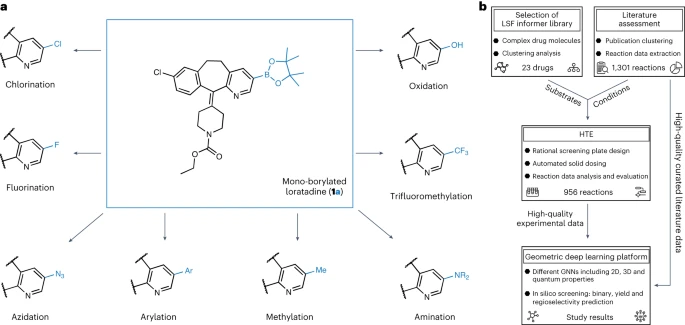
이 차트는 붕소화 다양화 연구의 개요를 보여줍니다. (데이터 출처: 논문)
인공지능 기반 LSF(언어 지원 기능)
고처리량 실험(HTE)은 반자동 소형화 및 소량 배치 스크리닝이 가능한 확립된 반응 최적화 방법으로, 소수의 귀중한 빌딩 블록과 소모품을 사용하여 여러 변환을 동시에 재현 가능하게 실행합니다. 성공 및 실패 응답에 대한 고품질 데이터세트를 생성하는 FAIR(검색 가능성, 접근성, 상호 운용성, 재사용성) 문서와 결합된 HTE는 고급 데이터 분석 및 기계 학습을 통해 약물 발견을 위한 LSF를 실현합니다.
GNN(Graph Neural Network)은 분자 특징 추출 및 속성 예측에 널리 사용되었습니다. 화학 반응 계획을 위해 개발된 다양한 기계 학습 방법 중에서 GNN은 역합성 계획, 위치 선택성 예측 및 반응 생성물 예측에 성공적으로 적용되었습니다. 또한 비슷한 문제를 해결하기 위해 변환기와 지문 기반 방법도 개발되었습니다.
연구에 따르면 전이 상태의 기하학을 학습하면 경쟁 반응의 결과를 정확하게 예측할 수 있는 것으로 나타났습니다. 밀도 범함수 이론(DFT)과 원자 부분 전하의 그래픽 특성화를 사용하면 전자 효과에 의해 구동되는 반응의 위치 선택성에 대한 예측이 향상될 수 있습니다. 그래프 기계 학습과 고처리량 실험(HTE)을 결합하여 유기 기판의 C-H 활성화 반응 조건을 최적화할 수 있습니다. 일부 연구에서는 거울상 선택성을 포함하여 반응 결과를 예측할 수 있는 전이 상태의 딥 러닝 모델 사용에 초점을 맞췄습니다.
그러나 이러한 방법은 작은 분자 구조와 상대적으로 작은 데이터 세트로 제한되어 이러한 모델을 구조적으로 더 복잡한 약물 유사 분자에 적용합니다. 문헌 연구를 기반으로 이리듐 촉매 보릴화 반응의 위치 선택성은 전이 상태의 양자 화학 정보로 강화된 하이브리드 기계 학습 모델을 통해 예측할 수 있습니다. 그러나 C-H 활성화 반응 모델의 성능과 다중 방향족 고리 시스템을 가진 분자에서의 위치 선택적 적용에 대한 입체 효과와 전자 효과의 영향은 아직 연구되지 않았습니다
기하학적 딥러닝을 사용한 자동화된 LSF 붕소화 스크리닝
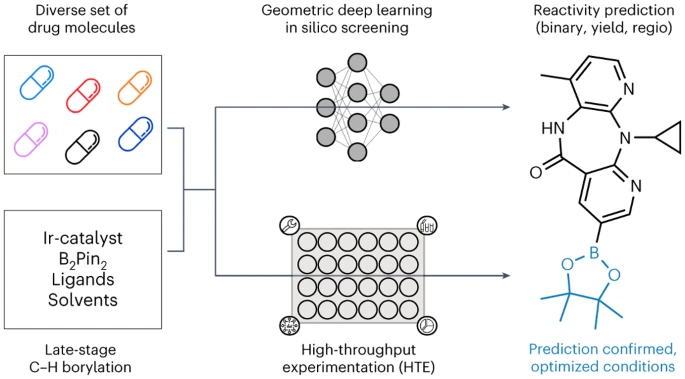
뮌헨 대학, ETH Zurich, Roche Pharmaceuticals Basel의 연구원들은 후기 단계의 히트 및 리드 다양화 기회를 식별하기 위해 기하학적 딥러닝에 적용되는 자동화된 LSF 붕소화 스크리닝 방법을 소개합니다. 복잡한 약물 분자 LSF의 반응 결과, 수율 및 위치 선택성을 예측하기 위해 컴퓨터 딥 러닝을 사용했습니다.
LMU 화학 및 약학대학과 Roche 연구 그룹의 수석 저자이자 박사 과정 학생인 David Nippa는 이 접근 방식을 사용하면 실험실 실험 횟수를 크게 줄여서 효율성을 높일 수 있다고 말했습니다. 화학적 합성 및 지속 가능성
이 연구의 첫 번째 단계에서 우리는 후기 단계 약물 발견 선도 화합물의 특성과 관련된 적절한 고처리량 스크리닝 반응 조건 및 기질을 선택하기 위해 출판된 문헌을 철저히 분석했습니다. 우리는 수동으로 정리된 38개의 문헌 데이터 세트를 기반으로 반응 조건을 결정했습니다
LSF 기질의 선택은 1,174개의 승인된 약물에 대한 클러스터 분석 결과를 기반으로 하여 구조적으로 다른 23개의 약물 분자를 생성했습니다. 이 접근 방식을 통해 연구자들은 납 화합물 합성을 최적화하기 위해 이상적인 기질과 적용 가능성이 제한된 단편에만 의존하는 대신 "정보 라이브러리" 접근 방식에서 반응 조건 및 기질의 관련 사례를 사용할 수 있습니다.
두 번째 단계에서 연구자들은 세미 -데이터(실험 데이터세트)를 생성하기 위한 자동화된 고처리량 실험(HTE). 선택된 약물 분자 및 반응 조건의 반응 데이터는 후속 반응 결과의 기계 학습을 위한 고품질 데이터를 제공합니다
마지막으로 2차원, 3차원 및 원자 부분 전하를 사용하여 다양한 그래프 신경망(GNN) 모델을 훈련했습니다. 강화 분자 그래프를 사용하여 이원 반응 결과, 반응 수율 및 위치 선택성을 예측합니다. ETH Zurich의 박사과정 학생인 Kenneth Atz는 "흥미롭게도 출발 물질에 대한 2차원 화학 공식이 아닌 3차원 정보를 고려할 때 예측이 향상되었습니다."라고 말했습니다.
이 접근 방식은 위치를 식별하는 데 성공적으로 사용되었습니다. 추가적인 반응성 그룹이 도입될 수 있는 기존 활성 성분 내에서. 이는 연구자들이 알려진 제약 활성 성분의 새롭고 더욱 효과적인 변종을 더욱 신속하게 개발하는 데 도움이 될 것입니다.
논문 내용을 보려면 다음 링크를 클릭하십시오: https://www.nature.com/articles/s41557-023-01360-5
관련 보고서: https://techxplore.com/news/2023-11-인공 지능이 약물의 길을 열어줍니다.html
위 내용은 기하학적 딥러닝 방법을 사용하여 약물 분자 합성을 위한 최적 옵션을 예측하여 신약 발견의 길을 열었습니다.의 상세 내용입니다. 자세한 내용은 PHP 중국어 웹사이트의 기타 관련 기사를 참조하세요!
관련 기사
더보기- 포산 마을 수준 산업 단지가 다시 태어났습니다. 로봇 지능형 제조 도시의 위아래 계단이 상류와 하류이고 산업 단지가 산업 체인입니다.
- 대회를 통해 학습 및 테스트 결과를 홍보함으로써 사물 인터넷 산업이 고품질 개발을 달성할 수 있도록 돕습니다.
- 2023년 제1회 장강 삼각주 로봇 산업 체인 협력 개발 정상 포럼이 안후이성 우후에서 성공적으로 개최되었습니다.
- 로봇 ETF(159770): 4일 연속 순자본 유입을 유치하는 '휴머노이드 로봇의 혁신 및 개발에 대한 지침 의견'은 산업 발전 프로세스를 촉진할 수 있습니다.
- 로봇산업 : 미래 9대 주력산업 중 하나인 AI 시대의 차세대 핫분야!