
DeepMind, 양자 화학 계산을 위한 신경망 변형 Monte Carlo 개발

- PHPz원래의
- 2024-07-16 15:26:30581검색

DeepMind와 Imperial College London의 연구원들은 최근 개발된 기본 세트에 의존하지 않는 Fermionic Neural Network(FermiNet) 파동 함수를 사용하여 이 문제를 해결합니다. FermiNet은 다양한 정성적 양전자 결합 특성을 지닌 다양한 원자 및 소분자에서 매우 정밀하고 경우에 따라 최첨단의 기저 상태 에너지를 생성하는 것으로 밝혀졌습니다.
연구원들은 도전적인 비극성 벤젠 분자의 결합 에너지를 계산하고 실험 값과 좋은 일치를 발견했으며 명시적으로 상관된 가우스 파동 함수를 사용하여 얻은 것보다 더 유리한 소멸 속도를 얻었습니다. 결과는 신경망 파동 함수를 기반으로 한 방법의 일반적인 장점을 보여 주며 이를 표준 분자 해밀턴 이외의 시스템에 적용합니다.
관련 연구는 "양전자 화학을 위한 신경망 변형 몬테카를로"라는 제목으로 "Nature Communications" 6월 18일자에 게재되었습니다.
논문 링크: https://www.nature.com/articles/s41467-024-49290-1 많은 양의 양전자를 포착하기 위한 실험 설정의 발전으로 인해 양전자 결합 상태를 설명하는 보다 효율적인 계산 도구가 개발되었습니다. 반물질 기술 혁신을 가속화합니다.  양전자-분자 복합체의 바닥 상태 특성에 대한 양자 화학 계산은 어렵습니다. 가장 큰 어려움은 전자와 양전자 사이의 유착을 나타내기 위해 적절한 기초 세트를 사용하는 데 있습니다.
양전자-분자 복합체의 바닥 상태 특성에 대한 양자 화학 계산은 어렵습니다. 가장 큰 어려움은 전자와 양전자 사이의 유착을 나타내기 위해 적절한 기초 세트를 사용하는 데 있습니다.
여기서 연구자들은 최근 QMC를 위해 개발된 신경망 파동 함수 가정을 기반으로 분자의 양전자 결합 상태의 바닥 상태 특성을 계산하는 새로운 방법을 제안합니다. 페르미온 신경망(FermiNet)은 일련의 기본 함수를 참조하지 않고 다물체 파동 함수를 모델링합니다. 이는 양전자 파동 함수를 설명할 때 위에서 설명한 많은 어려움을 편리하게 우회합니다.
연구원들은 FermiNet을 확장하여 파동 함수의 양전자 구성 요소를 전자 구성 요소와 동일하게 표현했습니다. 유연하고 정확한 하이브리드 전자-양전자 파동 함수 가정은 신경망 아키텍처를 최소한으로 변경하여 얻을 수 있습니다. 서로 다른 양전자 결합 메커니즘을 갖는 일련의 시스템에 대해 양전자 결합 에너지 및 소멸 속도를 계산했으며, 이러한 시스템의 바닥 상태 에너지에 대해 가장 높은 정확도를 얻었습니다.
양전자수소화물, 나트륨 및 마그네슘 원자, 작은 이원자 분자에 대한 결과는 이 방법이 이전 연구에 비해 최첨단 정확도를 달성할 수 있음을 보여줍니다. 또한, 비극성 디리튬 및 벤젠 분자에 대한 결과는 강한 전자-양전자 상관 효과에 의해 완전히 제어되는 양전자 결합 모드를 설명할 때 이러한 정밀도가 유지된다는 것을 보여줍니다.
아래 다이어그램은 비극성 분자와 양전자 사이의 결합 메커니즘에 대한 직관적인 이해를 제공합니다. 상관 관계 지배 결합은 분자 핵에서 멀어지는 증가된 전자 밀도 중심에 의해 촉진됩니다. 디리튬에서는 이것이 공유 결합이고, 벤젠에서는 고리 내 π 결합의 비편재화로 인해 분자 중심의 전자 밀도가 증가합니다.
그림: 양전자 리튬과 벤젠의 바닥 상태 단일 입자 밀도. (출처: 논문) 이 방법으로 얻은 벤젠의 양전자 결합 에너지는 Queen's University의 Hofierka et al.의 실험값 및 다체 이론과 매우 유사합니다. 그리고 얻은 소멸 속도는 알칼리 금속 원자 및 소분자의 고정밀 ECG-SVM 계산과 비슷합니다. 또한, FermiNet 파동함수를 이용하여 축적된 양전자 원자 및 분자의 2γ 소멸률을 다른 다양한 계산 방법으로 얻은 결과와 비교하면 다음 표와 같습니다.  표: 기타 다양한 계산 방법으로 얻은 소멸률과 비교 FermiNet 파동 함수를 사용하여 축적된 양전자 원자 및 분자의 비율, 2γ 소멸 속도입니다. (출처: 논문)
표: 기타 다양한 계산 방법으로 얻은 소멸률과 비교 FermiNet 파동 함수를 사용하여 축적된 양전자 원자 및 분자의 비율, 2γ 소멸 속도입니다. (출처: 논문)
1. 다른 방법과 비교하여 ECG-SVM은 파동함수를 가장 잘 포착하는 특징을 구성하여 소멸률을 계산합니다. 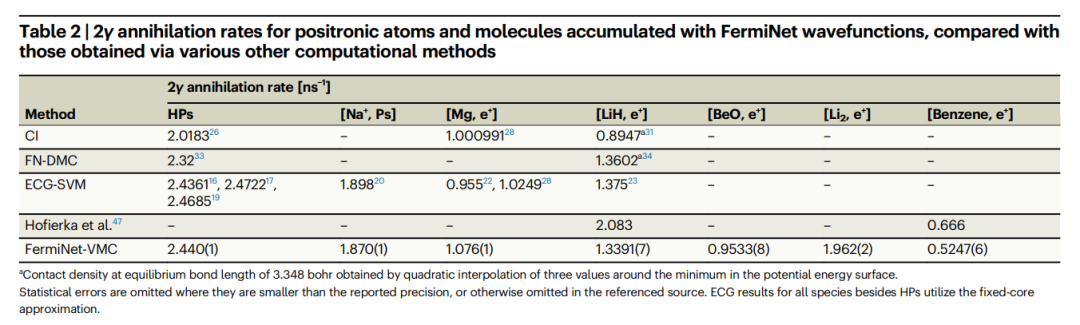
- 요약하자면, ECG-SVM 방법은 시스템별 조정 없이 다양한 양전자 결합 메커니즘을 통해 다양한 분자에 대해 매우 정확한 결과를 생성합니다.
위 내용은 DeepMind, 양자 화학 계산을 위한 신경망 변형 Monte Carlo 개발의 상세 내용입니다. 자세한 내용은 PHP 중국어 웹사이트의 기타 관련 기사를 참조하세요!