
Maison >Périphériques technologiques >IA >L'équipe de l'Université Jiao Tong de Shanghai développe un cadre d'apprentissage actif basé sur les données pour accélérer les progrès de la recherche sur les nanomatériaux de carbone
L'équipe de l'Université Jiao Tong de Shanghai développe un cadre d'apprentissage actif basé sur les données pour accélérer les progrès de la recherche sur les nanomatériaux de carbone

- WBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBavant
- 2024-01-16 22:33:191328parcourir
Éditeur |
Bien que la synthèse contrôlée de la croissance catalytique de substrat en nanostructures de carbone soit considérée comme une approche prometteuse, il existe encore des défis dans les mécanismes de croissance catalytique dynamique de surface et les stratégies de conception, qui nécessitent des recherches et développements supplémentaires.
Récemment, des équipes de recherche de l'Université Jiao Tong de Shanghai et de l'Université Tohoku au Japon ont démontré l'efficacité des modèles d'apprentissage automatique actifs pour révéler le processus microscopique de croissance catalytique du substrat. Grâce à l’application collaborative de la dynamique moléculaire et des méthodes de Monte Carlo, ils ont réalisé avec succès une simulation dynamique complète de la croissance du graphène sur Cu(111). Pour améliorer la précision de la simulation, l'équipe de recherche a adopté un potentiel d'approximation gaussienne. Cette recherche fournit de nouveaux outils et méthodes pour une compréhension approfondie du processus de croissance catalytique.
Grâce à cette recherche, nous avons développé une méthode pratique et efficace qui peut être utilisée pour concevoir des substrats métalliques ou en alliages afin d'obtenir les nanostructures de carbone souhaitées et d'explorer davantage de possibilités de réaction.
La recherche, intitulée « Modèle d'apprentissage automatique actif pour la simulation dynamique et les mécanismes de croissance du carbone sur une surface métallique », a été publiée dans « Nature Communications » le 6 janvier 2024.
 Lien papier : https://www.nature.com/articles/s41467-023-44525-z
Lien papier : https://www.nature.com/articles/s41467-023-44525-z
Le dépôt catalytique de substrat est considéré comme permettant d'obtenir un rendu bidimensionnel ou l’une des méthodes les plus prometteuses pour la croissance contrôlable de réseaux liés de manière covalente d’atomes de carbone tridimensionnels. Bien que les mécanismes de croissance sur les surfaces ordinaires aient été étudiés de manière approfondie, la connaissance des facteurs dynamiques et à l'échelle atomique qui contrôlent la masse du graphène sur les surfaces composites ou à indice élevé est limitée. Cette lacune en matière de recherche a grandement entravé le développement d’approches de conception guidées par la théorie pour de nouveaux substrats métalliques catalytiques dans la croissance de nanostructures de carbone.
La recherche expérimentale de catalyseurs métalliques ou en alliages présente des défis considérables en raison du large éventail de substrats potentiels et de la sensibilité du processus de croissance des nanomatériaux de carbone à divers paramètres expérimentaux.
Par conséquent, il y a suffisamment de place pour les simulations théoriques et de nombreux détails atomiques sont facilement obtenus. Les exemples incluent DFT, Kinetic Monte Carlo (KMC) et Ab initio Molecular Dynamics (AIMD). Cependant, ces méthodes ont chacune leurs limites. Par conséquent, il existe toujours un besoin urgent d’un modèle de conception robuste capable de décrire avec précision le mécanisme de croissance du carbone sur les surfaces métalliques.
Les potentiels d'apprentissage automatique (MLP) basés sur des réseaux de neurones artificiels ou des méthodes de noyau sont considérés comme des méthodes efficaces pour résoudre la précision et la transférabilité limitées des champs de force classiques et maintenir une précision de niveau DFT. Malgré des réalisations significatives dans les simulations MD basées sur les données, la construction de MLP précis reste une tâche difficile. Une solution à ce problème réside dans les techniques d’apprentissage dynamique.
Pour améliorer l'efficience et l'efficacité de la formation dynamique des processus de dépôt, un protocole de sélection bien défini est nécessaire. D’un autre côté, la dynamique de croissance du carbone sur les substrats métalliques peut être contrôlée par des événements rares et importants. Par conséquent, la manière d’améliorer l’efficacité de la formation du MLP en combinant des méthodes d’échantillonnage boosté avec la dynamique classique nécessite des recherches plus approfondies.
Cadre d'apprentissage automatique basé sur les données pour générer du MLP avec un minimum de main d'œuvreCette étude propose un cadre d'apprentissage automatique basé sur les données pour générer du MLP avec un minimum de main d'œuvre, adapté à la croissance du carbone sur des surfaces métalliques ou en alliages.
Pour réaliser cette tâche, les chercheurs ont utilisé (1) un modèle d'apprentissage du traitement du potentiel d'approximation gaussienne (GAP) ; (2) une méthode d'échantillonnage améliorée appelée Monte Carlo à biais de force horodatée (Monte Carlo à biais de force horodatée) ; Monte Carlo, tfMC) pour accélérer le processus de relaxation après le dépôt de carbone, incluant ainsi des événements rares et importants dans la base de données de formation (3) Sélectionner des données de formation représentatives basées sur le descripteur Smooth Overlap of Atomic Positions (SOAP) Stratégies efficaces (4) ; ensembles de formation sur le carbone bien établis ; (5) procédures automatisées de sélection, d’ajustement et de validation.
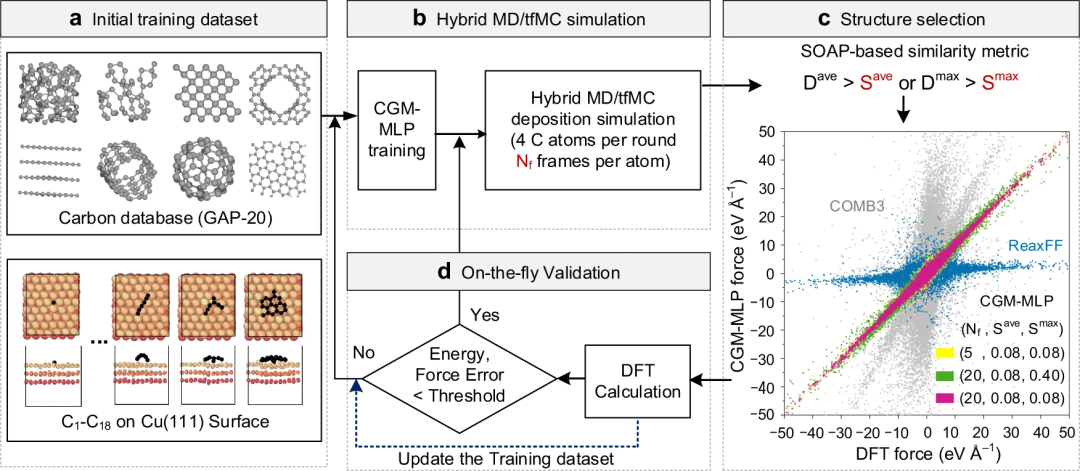 Figure 1 : Représentation schématique de la croissance du carbone sur le potentiel d'apprentissage automatique des métaux (CGM-MLP) générée par l'apprentissage actif dynamique lors d'une simulation hybride MD/tfMC. (Source : article)
Figure 1 : Représentation schématique de la croissance du carbone sur le potentiel d'apprentissage automatique des métaux (CGM-MLP) générée par l'apprentissage actif dynamique lors d'une simulation hybride MD/tfMC. (Source : article)
Réplication réussie de la nucléation du graphène et du carbone sur des surfaces métalliques en exploitant la haute précision du potentiel d'apprentissage automatique de croissance du carbone (CGM-MLP) et en incorporant des événements atomiques rares dans la méthode MD/tfMC. Les sous-processus de base liés à la croissance sont présentés dans la figure ci-dessous.
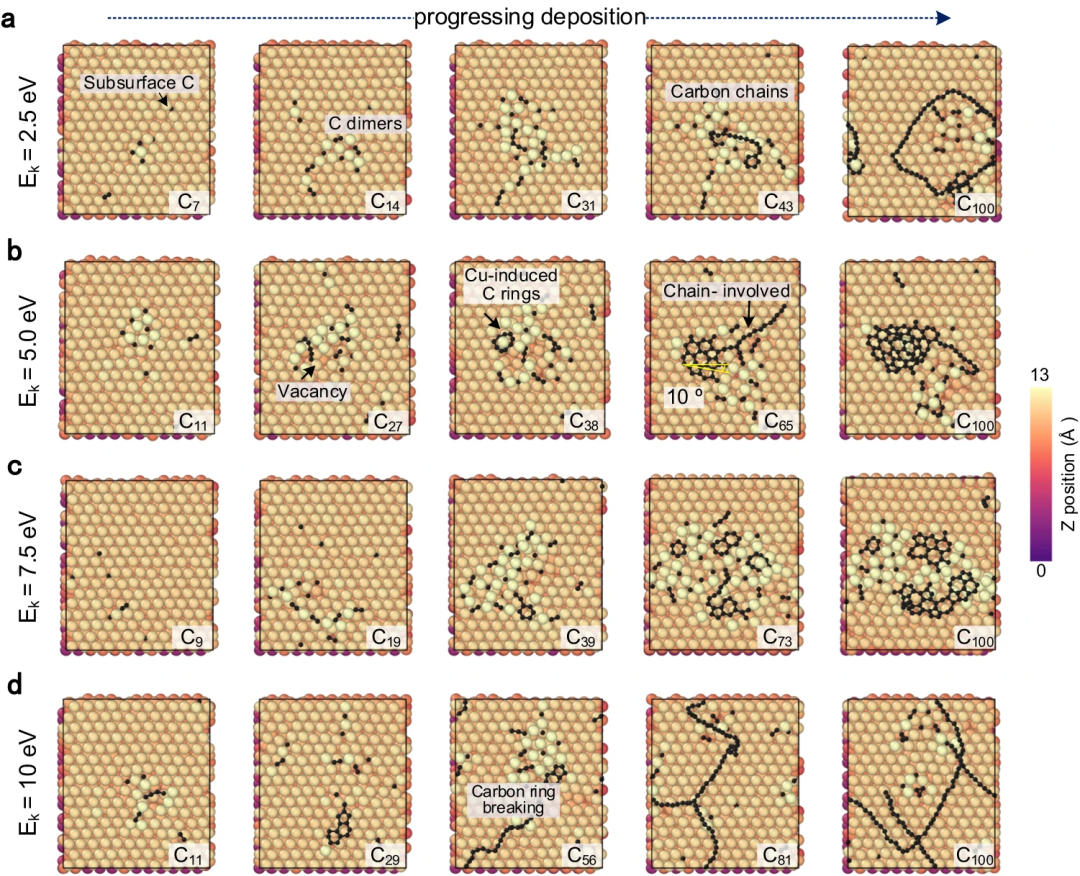
Figure 2 : Simulation pilotée par CGM-MLP de la croissance du graphène sur Cu(111) avec différentes énergies cinétiques incidentes du carbone (Ek). (Source : article)
Le potentiel résultant a ensuite été appliqué pour étudier la croissance du dépôt d'atomes de carbone sur la surface du Cu (111). Cette méthode peut capturer correctement les processus clés de la croissance du carbone sur Cu(111), tels que la formation et la migration de monomères de carbone souterrains et de dimères de surface, l'émergence de nanocristaux de carbone unidimensionnels, la nucléation du graphène impliquant des atomes de Cu et la passivation des bords du carbone. chaînes et processus de croissance des précipitations.
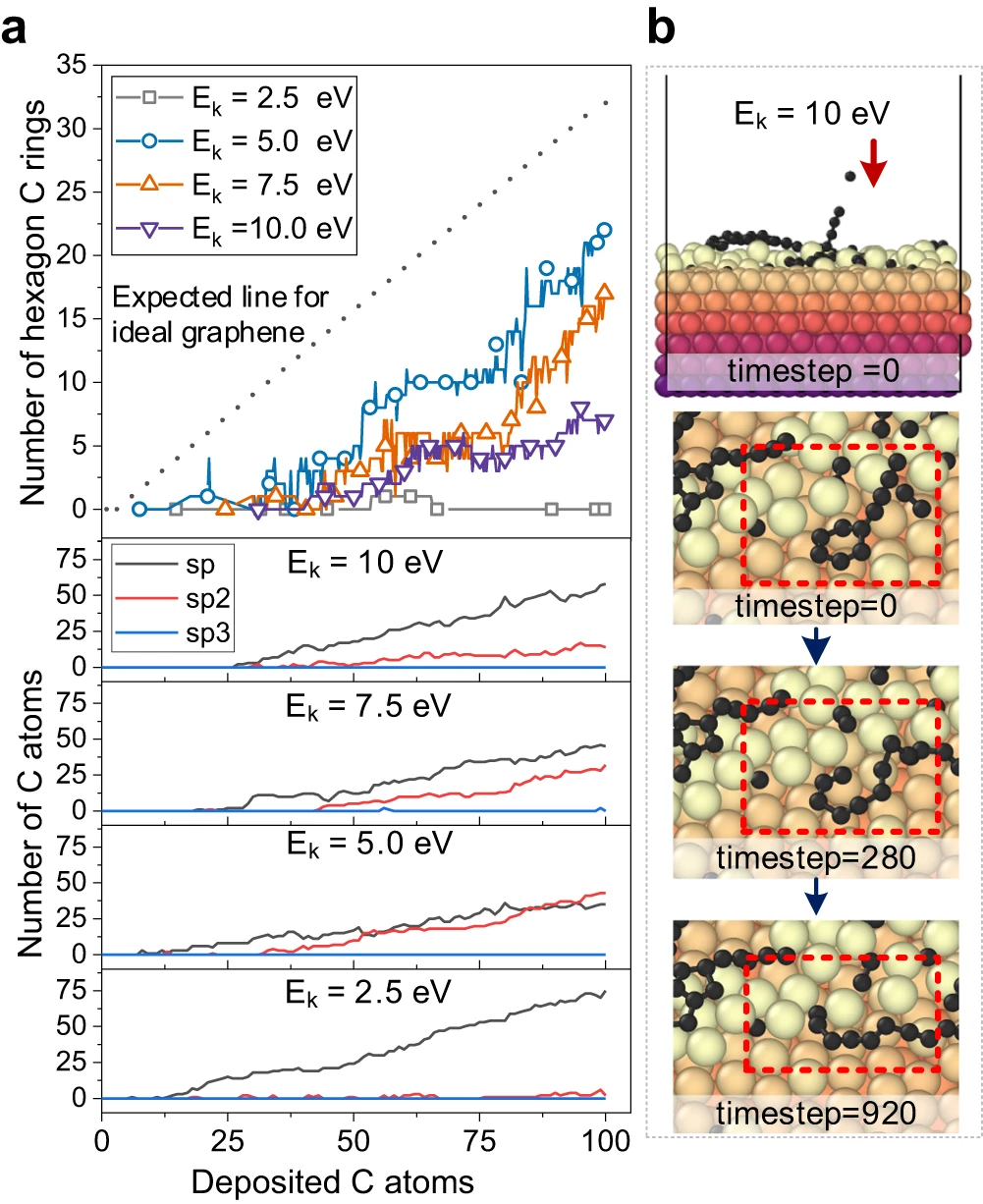
Figure 3 : Analyse de la structure du carbone et observation de la rupture des anneaux de carbone par bombardement à haute énergie. (Source : Papier)
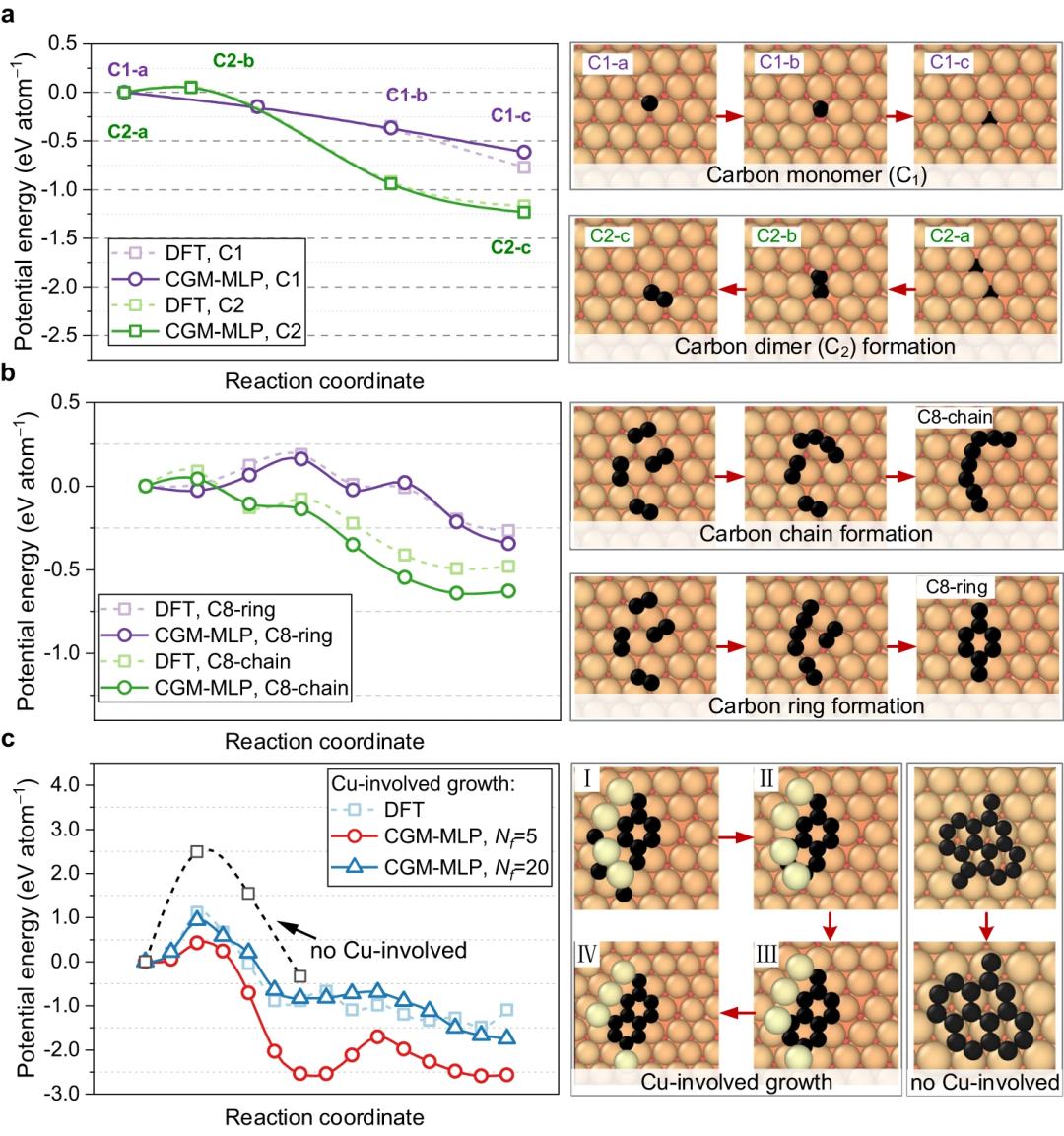
Figure 4 : Chemins d'énergie minimum pour la diffusion du carbone et la nucléation du graphène obtenus à l'aide de CGM-MLP sur du métal et de calculs de bande élastique poussée par image rampante (CI-NEB) basés sur DFT. (Source : article)
Les chercheurs ont simulé la nucléation initiale sur différentes surfaces métalliques, en particulier le dépôt de carbone sur Cu(111), Cr(110), Ti(001) et Cu(111) contaminé par l'O, avec des observations expérimentales et des calculs DFT faire preuve de cohérence.
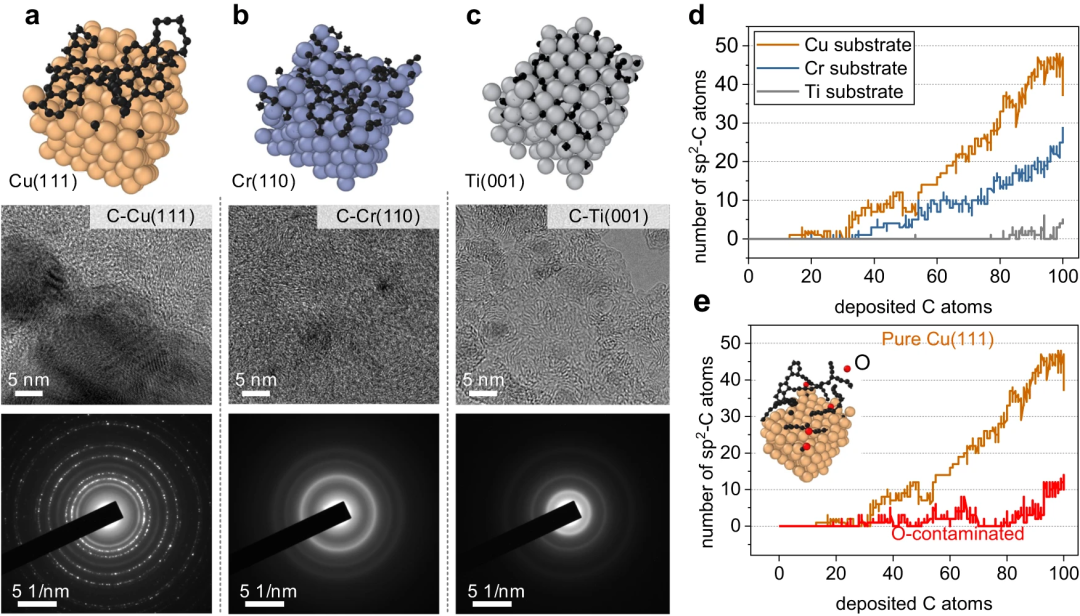
Figure 5 : Surfaces métalliques représentatives pour la croissance des nanostructures de carbone. (Source : article)
Importance de la recherche
En résumé, cette recherche représente une avancée pionnière dans l'intégration du MLP et du MD/tfMC, fournissant des informations transférables pour la conception de substrats métalliques ou en alliages afin d'obtenir les nanostructures de carbone souhaitées et une stratégie efficace.
CGM-MLP combine efficacement la précision des méthodes des premiers principes avec l'efficacité des champs de force classiques. La méthode tfMC surmonte les limitations d’échelle de temps des méthodes AIMD traditionnelles ou MD classiques. De plus, le cadre de formation automatisé de CGM-MLP intègre des stratégies de requêtes spécialisées pour créer des ensembles de formation dynamiques dans les simulations de dépôt, soulignant l'importance de prendre en compte l'environnement local autour des atomes déposés.
Ces avancées permettent d’étudier directement le mécanisme de croissance du carbone sur des surfaces métalliques complexes. Le modèle de dépôt basé sur l'apprentissage automatique proposé dans cette étude peut offrir la possibilité d'étudier la croissance d'une variété de nanostructures de carbone (par exemple, le graphène, les nanotubes de carbone, le graphite ou les films de carbone de type diamant) sur des substrats métalliques ou en alliages multi-éléments.
Ce qui précède est le contenu détaillé de. pour plus d'informations, suivez d'autres articles connexes sur le site Web de PHP en chinois!
Articles Liés
Voir plus- HTMLPurifier empêche les attaques XSS en PHP
- Parlons de la théorie de la réplication maître-esclave, de la sentinelle et du clustering dans Redis [explication détaillée avec images et texte]
- Parlons de reconnaissance d'image : réseau de neurones récurrents
- Que sont les réseaux de neurones convolutifs en Python ?