
Maison >Périphériques technologiques >IA >L'équipe Tsinghua propose un cadre de pré-formation sur les graphes guidés par les connaissances : une méthode pour améliorer l'apprentissage des représentations moléculaires
L'équipe Tsinghua propose un cadre de pré-formation sur les graphes guidés par les connaissances : une méthode pour améliorer l'apprentissage des représentations moléculaires

- WBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBavant
- 2023-11-23 18:17:071464parcourir

Pour faciliter la prédiction des propriétés moléculaires, il est très important d'apprendre une représentation efficace des caractéristiques moléculaires dans le domaine de la découverte de médicaments. Récemment, les gens ont surmonté le défi de la rareté des données en pré-entraînant les réseaux de neurones graphiques (GNN) à l'aide de techniques d'apprentissage auto-supervisées. Cependant, les méthodes actuelles basées sur l'apprentissage auto-supervisé présentent deux problèmes principaux : le manque de stratégies claires d'apprentissage auto-supervisé et les capacités limitées du GNN
Récemment, des équipes de recherche de l'Université Tsinghua, de l'Université de West Lake et du laboratoire Zhijiang ont proposé des connaissances. conseils Pré-formation guidée par les connaissances de Graph Transformer (KPGT), un cadre d'apprentissage auto-supervisé qui fournit des prédictions améliorées, généralisables et robustes des propriétés moléculaires grâce à un apprentissage de représentation moléculaire considérablement amélioré. Le framework KPGT intègre un transformateur de graphes conçu spécifiquement pour les graphes moléculaires et une stratégie de pré-formation guidée par les connaissances pour capturer pleinement les connaissances structurelles et sémantiques des molécules.
Grâce à des tests informatiques approfondis sur 63 ensembles de données, KPGT a démontré des performances supérieures dans la prédiction des propriétés moléculaires dans divers domaines. En outre, l'applicabilité pratique du KPGT dans la découverte de médicaments a été vérifiée en identifiant des inhibiteurs potentiels de deux cibles antitumorales. Dans l’ensemble, KPGT peut constituer un outil puissant et utile pour faire progresser le processus de découverte de médicaments assisté par l’IA.
La recherche s'intitulait « Un cadre de pré-formation guidé par les connaissances pour améliorer l'apprentissage des représentations moléculaires » et a été publiée dans « Nature Communications » le 21 novembre 2023.

La détermination expérimentale des propriétés moléculaires nécessite beaucoup de temps et de ressources, et l'identification de molécules possédant les propriétés souhaitées est l'un des défis les plus importants de la découverte de médicaments. Ces dernières années, les méthodes basées sur l’intelligence artificielle ont joué un rôle de plus en plus important dans la prédiction des propriétés moléculaires. L'un des principaux défis des méthodes basées sur l'intelligence artificielle pour prédire les propriétés moléculaires est la caractérisation des molécules
Ces dernières années, les méthodes basées sur l'apprentissage profond sont apparues comme des outils potentiellement utiles pour prédire les propriétés moléculaires, principalement en raison de leur capacité à extraire automatiquement à partir de données d’entrée simples Capacité supérieure à caractériser efficacement. Notamment, diverses architectures de réseaux de neurones, notamment les réseaux de neurones récurrents (RNN), les réseaux de neurones convolutifs (CNN) et les réseaux de neurones graphiques (GNN), sont capables de modéliser des données moléculaires dans divers formats, allant des entrées moléculaires simplifiées au système d'entrée de ligne ( SMILES) aux images moléculaires et aux diagrammes moléculaires. Cependant, la disponibilité limitée des molécules marqueurs et l’immensité de l’espace chimique limitent leurs performances prédictives, en particulier lorsqu’il s’agit d’échantillons de données non distribués.
Grâce aux réalisations remarquables des méthodes d'apprentissage auto-supervisées dans les domaines du traitement du langage naturel et de la vision par ordinateur, ces techniques ont été appliquées pour pré-entraîner les GNN et améliorer l'apprentissage des représentations de molécules, obtenant ainsi des résultats substantiels dans les tâches de prédiction des propriétés moléculaires en aval. . Progrès
Les chercheurs émettent l'hypothèse que l'introduction de connaissances supplémentaires décrivant quantitativement les caractéristiques moléculaires dans un cadre d'apprentissage auto-supervisé peut efficacement relever ces défis. Les molécules possèdent de nombreuses caractéristiques quantitatives, telles que des descripteurs moléculaires et des empreintes digitales, qui peuvent être facilement obtenues avec les outils informatiques actuellement établis. L'intégration de ces connaissances supplémentaires peut introduire de riches informations sémantiques moléculaires dans l'apprentissage auto-supervisé, améliorant ainsi considérablement l'acquisition de représentations moléculaires sémantiquement riches.
De manière générale, les méthodes d'apprentissage auto-supervisées existantes s'appuient sur GNN comme modèle de base. Cependant, GNN a une capacité de modèle limitée. De plus, les GNN peuvent avoir des difficultés à capturer les interactions à longue portée entre les atomes. Et les modèles basés sur Transformer sont devenus un modèle révolutionnaire. Il se caractérise par un nombre croissant de paramètres et la capacité de capturer des interactions à longue portée, offrant une approche prometteuse pour modéliser de manière exhaustive les caractéristiques structurelles des molécules
Cadre d'apprentissage auto-supervisé KPGT
Dans cette étude, les chercheurs ont introduit Un cadre d'apprentissage auto-supervisé appelé KPGT est développé pour améliorer l'apprentissage de la représentation moléculaire et promouvoir ainsi les tâches de prédiction des propriétés moléculaires en aval. Le cadre KPGT se compose de deux composants principaux : un modèle de base appelé Line Graph Transformer (LiGhT) et une politique de pré-formation guidée par les connaissances. Le framework KPGT combine le modèle LiGhT haute capacité, spécialement conçu pour modéliser avec précision les structures de graphes moléculaires, et utilise une stratégie de pré-formation guidée par les connaissances pour capturer la structure moléculaire et les connaissances sémantiques.
L'équipe de recherche a utilisé environ 2 millions de dollars du L'ensemble de données ChEMBL29 Molécule, LiGhT a été pré-entraîné via une stratégie de pré-entraînement guidée par les connaissances
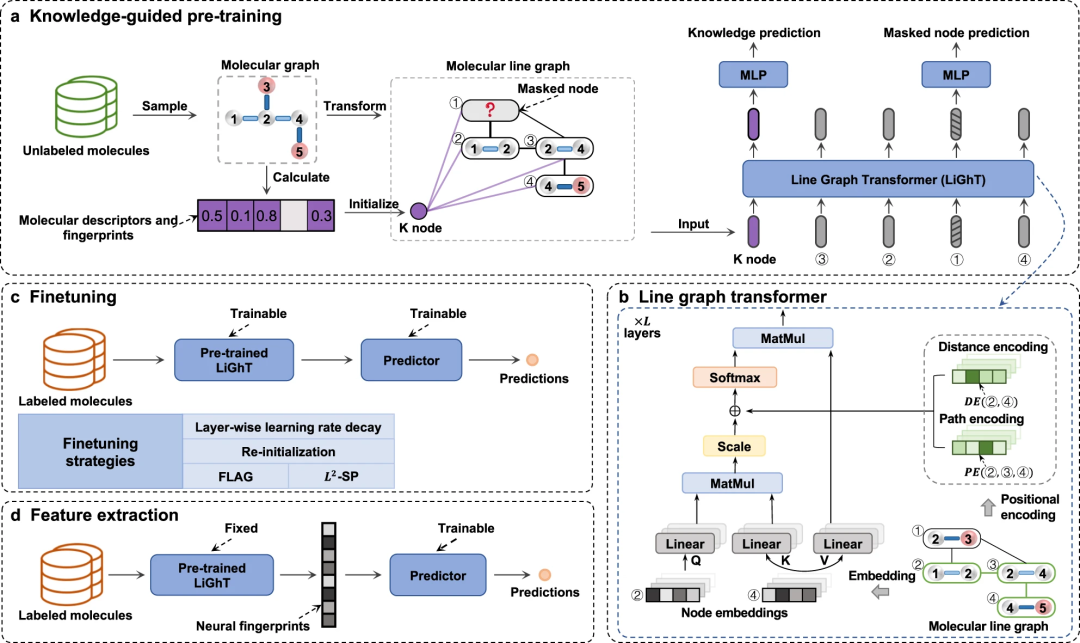
Contenu réécrit : Diagramme : Présentation de KPGT. (Source : article)
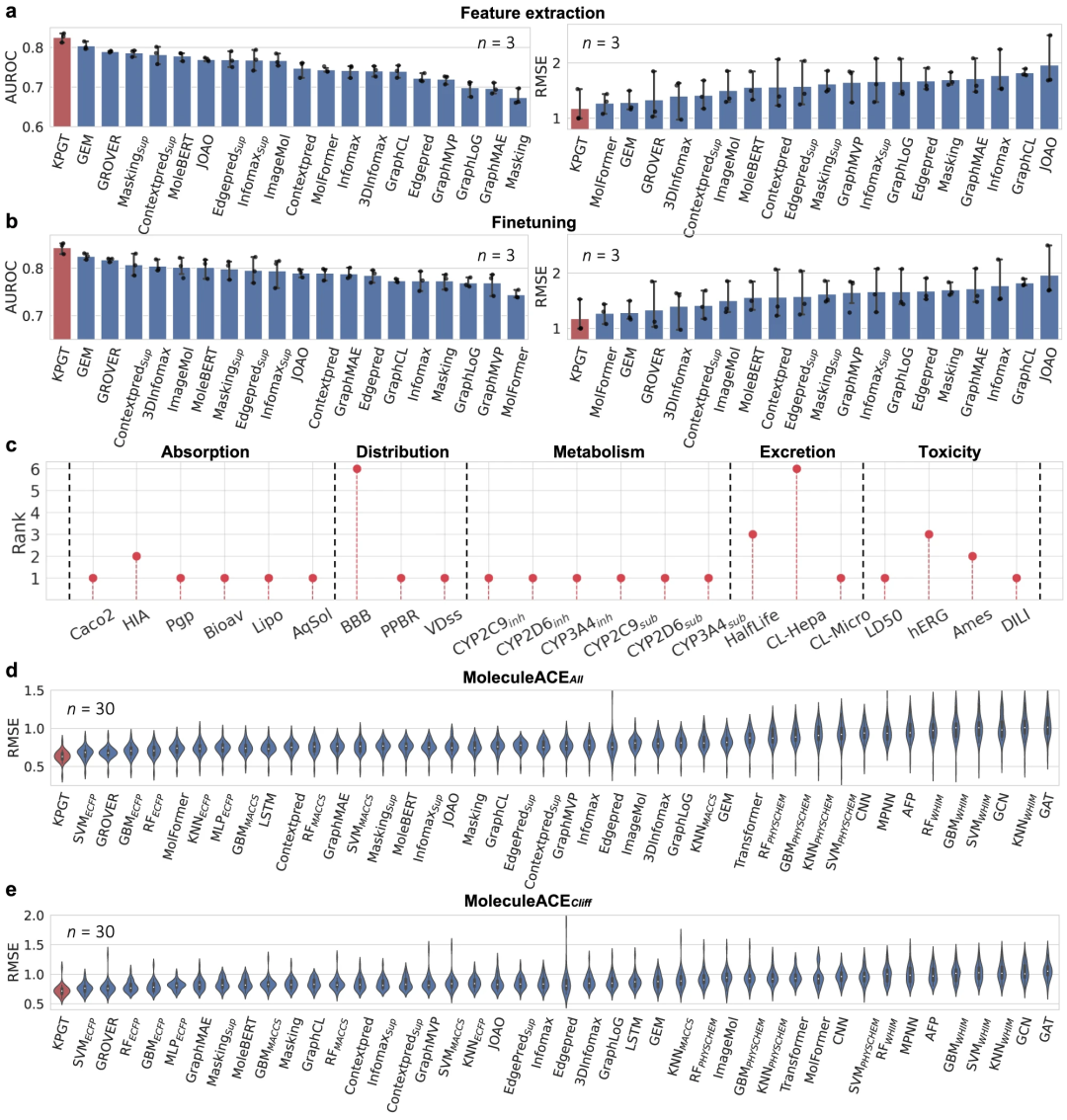
KPGT surpasse les méthodes de base en matière de prédiction des propriétés moléculaires. Par rapport à plusieurs méthodes de base, KPGT réalise des améliorations significatives sur 63 ensembles de données.
De plus, l'application pratique du KPGT a été démontrée en utilisant avec succès le KPGT pour identifier des inhibiteurs potentiels de deux cibles antitumorales, la kinase progénitrice hématopoïétique 1 (HPK1) et le récepteur du facteur de croissance des fibroblastes sexuels (FGFR1).
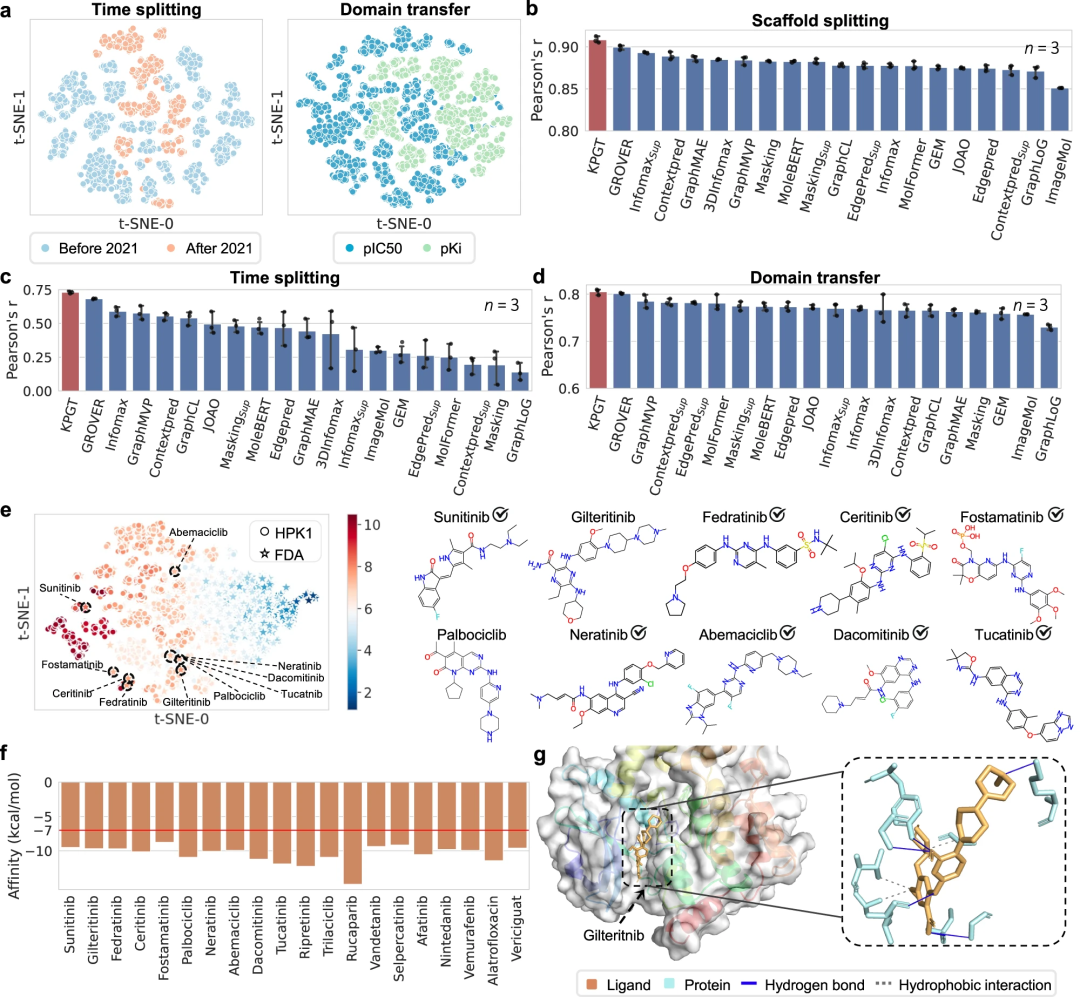
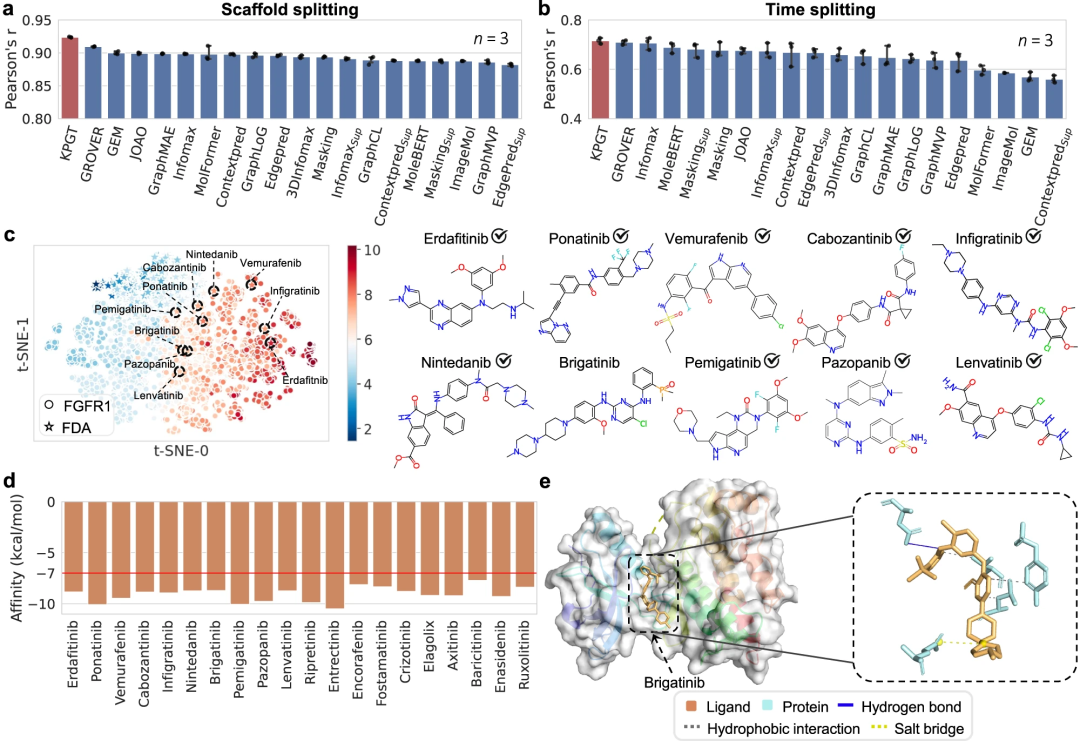
Limites de la recherche
Malgré les avantages du KPGT dans la prédiction efficace des propriétés moléculaires, il existe encore certaines limites.
Tout d'abord, l'intégration de connaissances supplémentaires est la caractéristique la plus significative de la méthode proposée. En plus des 200 descripteurs moléculaires et des 512 RDKFP utilisés dans KPGT, il est possible d'incorporer divers autres types de connaissances complémentaires. De plus, des recherches ultérieures pourraient intégrer des conformations moléculaires tridimensionnelles (3D) dans le processus de pré-entraînement, permettant au modèle de capturer des informations 3D importantes sur la molécule et d'améliorer potentiellement les capacités d'apprentissage des représentations. Bien que KPGT utilise actuellement un modèle de base avec environ 100 millions de paramètres et un pré-entraînement sur 2 millions de molécules, l'exploration d'un pré-entraînement à plus grande échelle pourrait offrir des avantages plus substantiels pour l'apprentissage des représentations moléculaires.
Dans l'ensemble, KPGT fournit un puissant cadre d'apprentissage auto-supervisé pour un apprentissage efficace des représentations moléculaires, faisant ainsi progresser le domaine de la découverte de médicaments assistée par l'intelligence artificielle.
Lien papier : https://www.nature.com/articles/s41467-023-43214-1
Ce qui précède est le contenu détaillé de. pour plus d'informations, suivez d'autres articles connexes sur le site Web de PHP en chinois!
Articles Liés
Voir plus- Tutoriel d'introduction au développement de la plateforme publique WeChat (explication détaillée avec images et textes)
- Explication détaillée du didacticiel d'introduction ABP de la série ABP du cadre de développement de modèles ASP.NET
- Quels livres dois-je lire pour apprendre Java à partir de zéro ? Livres Java avancés recommandés
- Ce qu'il faut apprendre en premier pour débuter avec la programmation
- Introduction aux bases du réseau