
Maison >Périphériques technologiques >IA >Un chercheur en médicaments IA rejoint la sous-revue Nature : utiliser ses connaissances professionnelles pour accélérer le développement de médicaments
Un chercheur en médicaments IA rejoint la sous-revue Nature : utiliser ses connaissances professionnelles pour accélérer le développement de médicaments

- 王林avant
- 2023-11-02 17:45:231254parcourir
La découverte de médicaments est un processus complexe en plusieurs étapes impliquant l'intersection de nombreuses sous-disciplines de la chimie et de la biologie. Les chimistes médicinaux humains jouent un rôle important dans ce processus grâce à leurs années d'expertise accumulées
Alors, l’intelligence artificielle (IA) peut-elle remplir le rôle que jouent les chimistes médicinaux dans la découverte de médicaments ? La réponse est peut-être oui.
Récemment, une équipe de recherche des Instituts Novartis pour la recherche biomédicale (NIBR) et du Centre de recherche Microsoft pour l'intelligence scientifique (AI4Science) a proposé conjointement un modèle d'apprentissage automatique capable de reproduire partiellement les connaissances collectives accumulées par les chimistes professionnels dans leur travail. , ce type de connaissance est souvent appelé « intuition chimique ».
L'équipe de recherche estime que cette méthode peut être utilisée en complément de la modélisation moléculaire pour améliorer l'efficacité du développement futur de médicaments
Le document de recherche s'intitule "Extracting Intuition in Medicinal Chemistry through Preference Machine Learning" et a été publié dans Nature Communications, une sous-revue de Nature

L'apprentissage automatique recrée l'expertise des chimistes médicinaux
Les chimistes médicinaux, qu'ils soient en laboratoire humide ou en informatique, jouent un rôle crucial dans la phase « d'optimisation des leads » de la découverte de médicaments, car on leur demande souvent de déterminer quels composés doivent être synthétisés et utilisés dans les cycles d'optimisation ultérieurs pour être évalués.
Pour ce faire, les chimistes médicinaux examinent généralement les données comprenant les propriétés des composés telles que l'activité, ADMET2 ou les informations structurelles cibles. Ainsi, le succès d’un projet dépend non seulement de la qualité des données expérimentales générées, mais également de la robustesse et de la rationalité des décisions prises par l’équipe travaillant en chimie médicinale.
Les chimistes médicinaux sont capables de prendre des décisions plus efficacement car ils s'appuient souvent sur leur expertise pour avoir une compréhension intuitive de ce qui réussit dans les différentes itérations de la découverte précoce de médicaments.
Bien qu'il y ait eu des tentatives antérieures pour formaliser ces connaissances à l'aide d'approches basées sur des règles ou de simples scores de faisabilité chimioinformatiques, capturer la subtilité et la complexité impliquées dans la notation par les chimistes médicinaux reste un défi fondamental
Pour atteindre cet objectif, la recherche vise à transformer l’expertise en une partie d’un modèle d’apprentissage automatique. Ce modèle peut être utilisé comme outil auxiliaire, comme d'autres systèmes de recommandation signalés dans l'industrie, pour déployer le processus de prise de décision dans l'optimisation des leads ou d'autres aspects de la découverte de médicaments
Considérant que la chimie médicinale repose actuellement principalement sur le travail manuel, elle est inévitablement affectée par des biais subjectifs. Certaines études ont rapporté un faible accord dans les évaluations entre les chimistes médicinaux ainsi qu'au sein des chimistes médicinaux. Dans cette étude, les chercheurs espèrent résoudre certains problèmes en empruntant des stratégies aux jeux multijoueurs.
Ils ont traité la tâche de classement d'un ensemble de molécules comme un problème d'apprentissage des préférences, puis ont utilisé un simple réseau neuronal pour modéliser les préférences individuelles
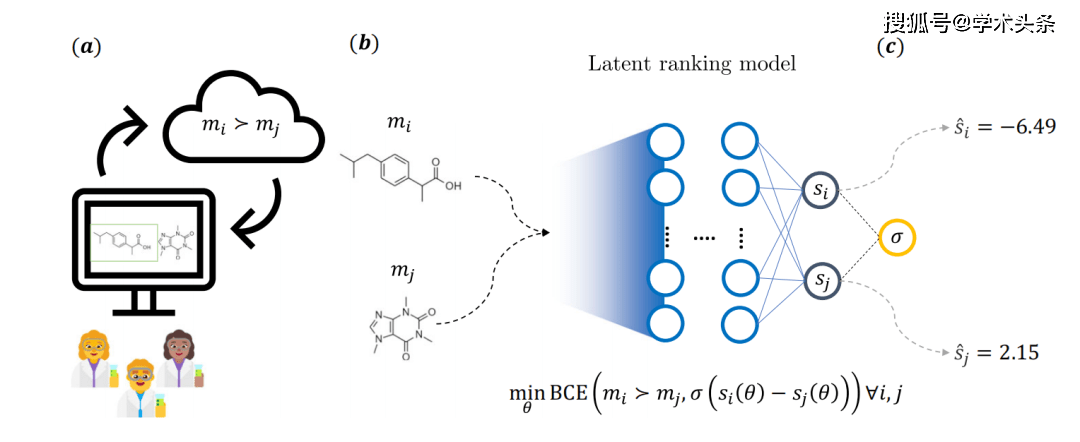 Figure | Schéma général des idées principales de la recherche (source : l'article)
Figure | Schéma général des idées principales de la recherche (source : l'article)
Plus précisément, comme le montre la figure ci-dessus, les molécules sont considérées comme des participants à un jeu compétitif, la probabilité qu'un camp gagne étant déterminée par les commentaires fournis par le chimiste. Pour ce faire, les chimistes médicinaux répondent à des questions prédéfinies sur une application Web et sélectionnent l’une des deux molécules. Au total, 35 chimistes médicinaux de Novartis ont été impliqués dans le processus, qui a abouti à la collecte de plus de 5 000 annotations.
Ces retours ont conduit à un modèle de notation implicite, qui utilise un modèle avec deux structures de réseaux neuronaux indépendantes. Chaque branche a un poids fixe et les molécules sont caractérisées à l'aide de descripteurs chimioinformatiques communs. Pendant la formation, les paramètres du modèle sont optimisés via une perte d'entropie croisée binaire (perte BCE), qui dépend de la différence de score sous-jacente d'une paire de molécules et des commentaires fournis par le chimiste
Une fois la formation terminée, le score de toute molécule arbitraire peut être déduit, qui peut ensuite être utilisé pour des tâches cheminformatiques en aval.
De plus, le modèle peut déterminer plus précisément les similitudes entre les différents médicaments. La fonction de notation d'apprentissage proposée dans l'étude est plus précise que l'indice d'évaluation de similarité des médicaments (QED) traditionnel
Notamment, Pour favoriser la reproductibilité de l'étude et le développement ultérieur du domaine, les chercheurs fournissent également un progiciel appelé "MolSkill" qui contient le modèle et les données de réponse anonymisées.
Problèmes et applications de l'apprentissage automatique dans le domaine de la chimie médicinale
Cependant, bien que ce modèle puisse reproduire les connaissances accumulées par les chimistes médicinaux dans leur travail, il présente également certaines limites. Premièrement, afin de capter l'intuition chimique, les questions posées lors de la collecte de données ont toujours été vagues.
De plus, bien que le plan d'étude proposé ait abouti à un plus grand accord entre les participants par rapport aux études précédentes, la méthode de comparaison par paires n'est pas parfaite.
De plus, le « Flatland Fallacy » conduit les humains à tendre à simplifier les problèmes de grande dimension en un petit ensemble de variables qui peuvent être suivies cognitivement, et cette simplification peut être affectée par les caractéristiques personnelles de chaque chimiste médicinal
Cependant, l'équipe de recherche a déclaré que le modèle proposé dans cette étude ne se limite pas au champ d'application de l'étude actuelle. Plus précisément, le cadre discuté peut être étendu à d'autres observables quantifiables mais coûteux dans le domaine de la découverte de médicaments. En outre, cela peut fournir un aperçu de domaines encore inexplorés de l’espace chimique.
Dans cet esprit, l'équipe de recherche estime qu'une architecture similaire peut être construite en permettant à certains filtres basés sur des règles populaires d'apprendre à partir de données d'entraînement générées artificiellement. Ce modèle peut surmonter la limitation majeure liée à la nécessité de filtrer manuellement les composés avant de faire des inférences
La même approche peut également être utilisée pour générer des scores composés en donnant la priorité aux combinaisons dans des bibliothèques chimiques synthétiques où le criblage en raison de leur nouveauté naturelle est difficile à l'aide des méthodes basées sur des règles existantes
Une autre chose qui doit être réexprimée est la suivante : dans un scénario prospectif d'optimisation primaire pour une cible spécifique, plusieurs sources d'informations (telles que les propriétés biologiques, ADMET, etc.) doivent être prises en compte de manière globale pour tester le caractère pratique du cadre de recherche
L'équipe de recherche a écrit dans l'article : « Les méthodes d'apprentissage automatique peuvent concevoir des milliers de composés, et des technologies telles que le criblage à haut débit peuvent mettre en évidence un grand nombre de composés candidats dès les premières étapes du processus de découverte de médicaments. le temps est venu. On s'attend à ce que cette application accélère l'adoption de la méthode et la confiance dans les années à venir »
.Ce qui précède est le contenu détaillé de. pour plus d'informations, suivez d'autres articles connexes sur le site Web de PHP en chinois!