
Maison >Périphériques technologiques >IA >Meta AI ouvre plus de 600 millions de cartes de structure de protéines métagénomiques et 15 milliards de modèles de langage ont été réalisés en deux semaines
Meta AI ouvre plus de 600 millions de cartes de structure de protéines métagénomiques et 15 milliards de modèles de langage ont été réalisés en deux semaines

- 王林avant
- 2023-04-16 11:37:021752parcourir
Cette année, DeepMind a publié les structures prédites d'environ 220 millions de protéines, ce qui couvre presque toutes les protéines des organismes connus dans la base de données ADN. Aujourd’hui, un autre géant de la technologie, Meta, comble un autre vide, celui des microbes.
En termes simples, Meta utilise la technologie de l'IA pour prédire les structures d'environ 600 millions de protéines provenant de bactéries et d'autres micro-organismes encore à caractériser. Le chef d'équipe, Alexander Rives, a déclaré : « Ces protéines sont les structures que nous connaissons le moins, et ce sont des protéines très mystérieuses. Je pense que ces découvertes offrent le potentiel d'une compréhension approfondie de la biologie.
Habituellement, les modèles de langage sont utilisés dans. » grandes quantités Formé sur le texte. Meta Pour appliquer des modèles de langage aux protéines, Rives et ses collègues ont pris comme entrée des séquences protéiques connues, composées de 20 acides aminés représentés par des lettres différentes. Le réseau a alors appris à auto-compléter les protéines tout en masquant une certaine proportion d’acides aminés.
Meta a nommé ce réseau ESMFold. Bien que la précision de prédiction d'ESMFold ne soit pas aussi bonne que celle d'AlphaFold, elle est environ 60 fois plus rapide que celle d'AlphaFold pour prédire les structures. Cette vitesse signifie que les prédictions de la structure des protéines peuvent être étendues à des bases de données plus grandes.

- Adresse de papier: https://www.biorxiv.org/content/10.1101/2022.07.20.500902v2
- project Adresse: https://github.com/facebookresearch/esm
Maintenant, à titre de test, Meta a décidé d'appliquer son modèle à une base de données d'ADN métagénomique, tous provenant de l'environnement, y compris le sol, l'eau de mer, l'intestin humain, la peau et d'autres habitats microbiens. Meta AIannonce le lancement de l'Atlas métagénomique ESM contenant plus de 600 millions de protéines, qui constitue la première vue complète de la « matière noire » de l'univers des protéines. Il s’agit également de la plus grande base de données de structures prédites à haute résolution, 3 fois plus grande que n’importe quelle base de données existante sur la structure des protéines, et la première à fournir une couverture complète et à grande échelle des protéines métagénomiques.
Au total, l'équipe Meta a prédit plus de 617 millions de structures protéiques en seulement deux semaines. Rives a déclaré que les prédictions sont gratuites et accessibles à tous, tout comme le code sous-jacent du modèle.
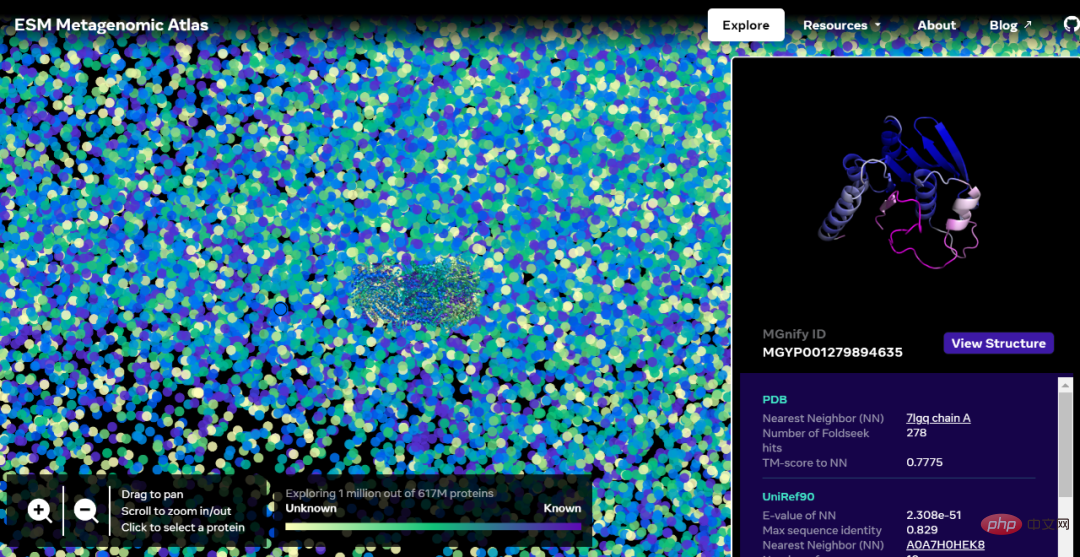
Adresse de la version interactive : https://esmatlas.com/explore?at=1%2C1%2C21.999999344348925
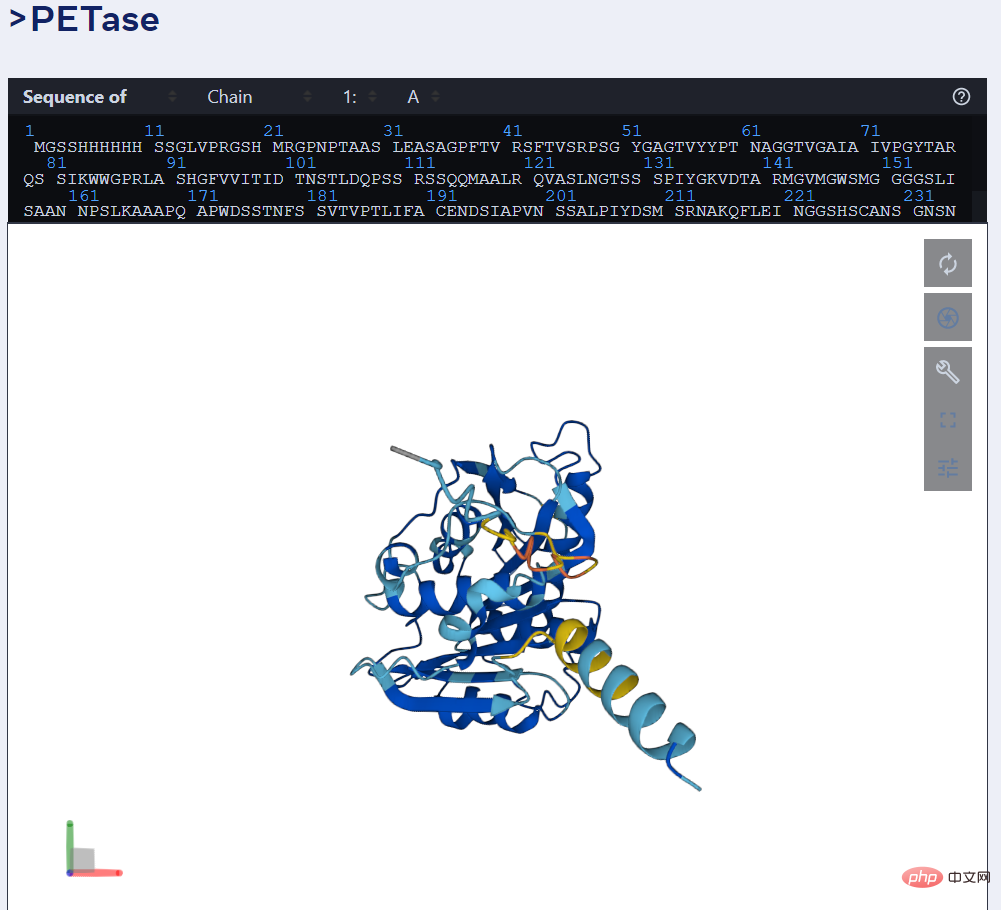
Par exemple, l'image ci-dessous montre la prédiction d'ESMFold sur l'enzyme PET.
Introduction
Comme nous le savons tous, les protéines sont des molécules complexes et dynamiques codées par des gènes et sont principalement responsables des processus fondamentaux de la vie. Les protéines jouent un rôle étonnant en biologie. Par exemple, les bâtonnets et les cônes de l’œil humain détectent la lumière afin que nous puissions voir le monde extérieur ; les capteurs moléculaires qui constituent la base de l’audition et du toucher ; les molécules complexes des plantes qui convertissent l’énergie lumineuse en énergie chimique ; les micro-organismes et les « moteurs » qui font bouger les muscles humains ; les enzymes qui décomposent le plastique ; les anticorps qui nous protègent des maladies, etc. sont tous des protéines.
En 1998, Jo Handelsman du Département de phytopathologie de l'Université du Wisconsin a proposé pour la première fois le concept de métagénomique, qui provient dans une certaine mesure de l'étude et de l'analyse d'ensembles de gènes de l'environnement comme un seul génome. deet macro en anglais est méta-, qui est également traduit par yuan.
Metagenomics révèle des milliards de séquences protéiques nouvelles pour la science, cataloguées pour la première fois par le NCBI (European Bioinformatics Institute et Joint Genome Institute) et d'autres grandes bases de données compilées par des projets publics.
Nouvelle méthode de repliement des protéines développée par Meta AI qui exploite de grands modèles de langage pour créer la première vue complète de la structure des protéines dans des bases de données métagénomiques (avec des centaines de millions de protéines). Meta a découvert que les modèles de langage peuvent prédire la structure tridimensionnelle des protéines au niveau atomique 60 fois plus rapidement que les méthodes existantes de prédiction de la structure des protéines SOTA. Cette avancée contribuera à accélérer une nouvelle ère de compréhension de la structure des protéines, permettant pour la première fois de comprendre les structures des milliards de protéines cataloguées par la technologie de séquençage génétique.
Déverrouiller le monde caché de la nature : la première vue complète de l'espace structurel métagénomique
Nous savons que les progrès du séquençage génétique ont permis de cataloguer des milliards de séquences protéiques métagénomiques. Mais la détermination expérimentale de la structure 3D de milliards de protéines va bien au-delà des techniques de laboratoire chronophages telles que la cristallographie aux rayons X, qui peuvent prendre des semaines, voire des années, pour détecter une seule protéine. Les approches informatiques peuvent fournir des informations sur les protéines métagénomiques qui ne sont pas possibles à l’aide de techniques expérimentales.
La cartographie métagénomique ESM permettra aux scientifiques de rechercher et d'analyser la structure des protéines métagénomiques à l'échelle de centaines de millions de protéines. Cela peut aider à identifier des structures jusqu’alors non caractérisées, à rechercher des relations évolutives distantes et à découvrir de nouvelles protéines qui pourraient être utilisées en médecine et dans d’autres applications.
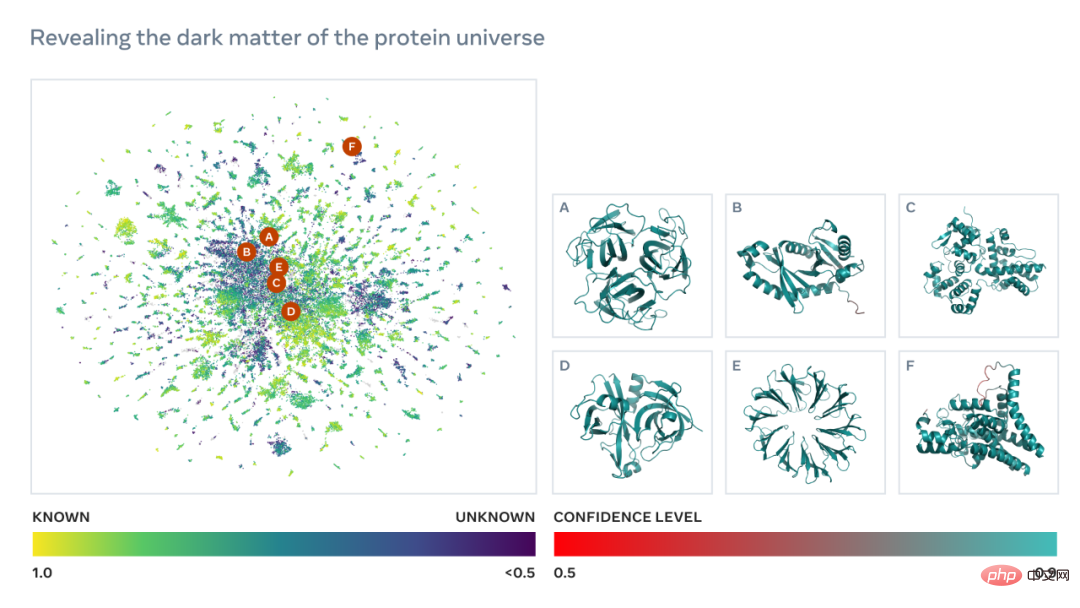
Ce qui suit est une carte contenant des dizaines de milliers de prédictions de haute confiance, montrant des similitudes avec des protéines dont les structures sont actuellement connues. Et, pour la première fois, l’image montre une région beaucoup plus grande de l’espace de structure des protéines qui était complètement inconnue.
Apprenez à lire le langage biologique
Comme le montre la figure ci-dessous, le modèle de langage ESM-2 est entraîné pour prédire les acides aminés qui ont été masqués par séquence au cours de l'évolution. Meta AI a découvert que, suite à la formation, des informations sur la structure des protéines apparaissaient dans l’état interne du modèle. C'est surprenant puisque le modèle n'a été entraîné que sur des séquences.
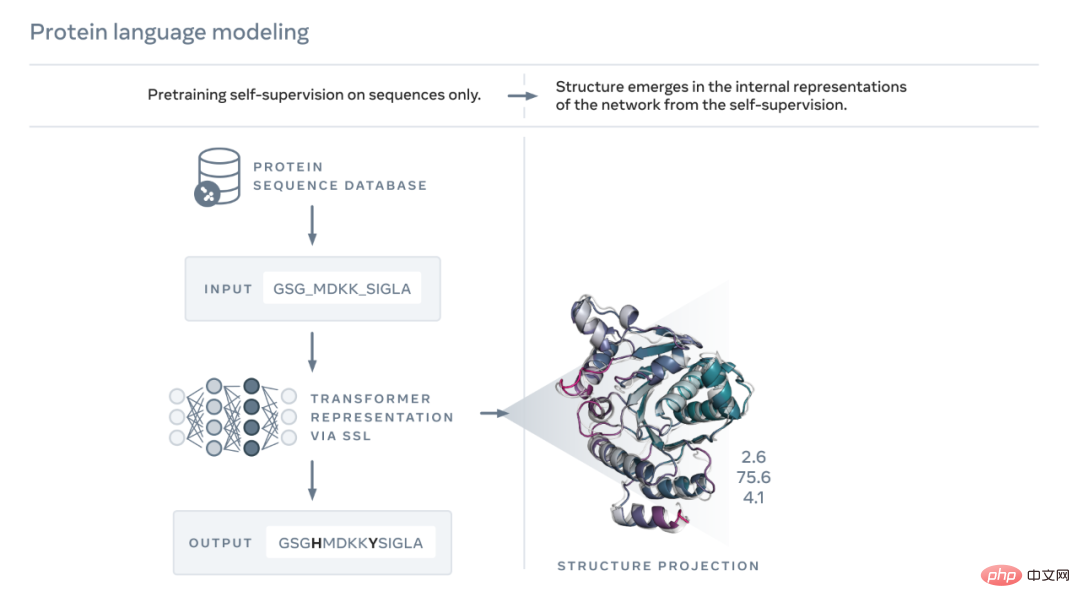
Tout comme le texte d'un papier ou d'une lettre, les protéines peuvent être écrites sous forme de séquences de caractères. Chaque caractère correspond à l'un des 20 éléments chimiques standards (acides aminés), chacun ayant des propriétés différentes et qui sont les éléments constitutifs des protéines. Ces éléments constitutifs peuvent être assemblés de manières astronomiquement différentes, par exemple pour une protéine composée de 200 acides aminés, il existe 20^200 séquences possibles, ce qui est plus que le nombre d'atomes dans l'univers visible. Chaque séquence se plie en une forme 3D (mais toutes les séquences ne se plient pas en une structure cohérente, beaucoup se plient en une forme désordonnée), et c'est cette forme qui détermine en grande partie la fonction biologique de la protéine.
Apprendre à lire le langage biologique comporte de grands défis. Bien que les séquences de protéines et les passages de texte puissent être écrits sous forme de caractères, il existe des différences profondes et fondamentales entre eux. Une séquence protéique décrit la structure chimique d’une molécule qui se plie en formes 3D complexes selon les lois de la physique.
Les séquences protéiques contiennent des modèles statistiques qui transmettent des informations sur la structure de repliement des protéines. Par exemple, si deux positions dans une protéine coévoluent, ou en d’autres termes, si un certain acide aminé apparaît à une position et s’associe généralement à un certain acide aminé à l’autre position, cela peut signifier que les deux positions sont dans la structure repliée. interaction. Cela ressemble à deux pièces d’un puzzle, où l’évolution doit choisir les acides aminés qui s’emboîtent dans une structure pliée. Cela signifie que nous pouvons souvent déduire la structure d’une protéine en observant des modèles dans sa séquence.
ESM utilise l'IA pour apprendre à lire ces modèles. En 2019, Meta AI a prouvé que les modèles de langage apprennent les propriétés des protéines, telles que leur structure et leur fonction. Grâce à une forme d’apprentissage auto-supervisé appelée modélisation du langage masqué, Meta AI a formé un modèle de langage sur les séquences de millions de protéines naturelles. Grâce à cette méthode, le modèle doit combler correctement les lacunes du paragraphe de texte, telles que "To _ or not to , c'est le _____".
Ensuite, Meta AI entraîne un modèle de langage pour combler les lacunes de la séquence protéique. Ils ont découvert que des informations sur la structure et la fonction des protéines apparaissaient au cours de cette formation. En 2020, Meta a publié un modèle de langage protéique SOTA, ESM1b, pour diverses applications, notamment pour aider les scientifiques à prédire l’évolution du COVID-19 et à découvrir les causes génétiques de la maladie.
Maintenant, Meta AI a étendu cette approche pour créer le modèle de langage protéique de nouvelle génération ESM-2, qui, avec 15 milliards de paramètres, est le plus grand modèle de langage protéique à ce jour. Ils ont constaté que lorsque les paramètres du modèle passaient de 8 millions à 15 milliards, des informations apparaissaient dans la représentation interne, permettant des prédictions de structure 3D à une résolution atomique.
Obtenez une accélération de plusieurs ordres de grandeur dans le repliement des protéines
Dans la figure ci-dessous, à mesure que le modèle est agrandi, une structure protéique à haute résolution apparaît. En même temps, à mesure que le modèle évolue, de nouveaux détails apparaissent dans les images à résolution atomique de la structure des protéines.

En utilisant les outils informatiques SOTA actuels, prédire la structure de centaines de millions de séquences protéiques sur une échelle de temps réaliste pourrait prendre des années, même avec les ressources des grandes institutions de recherche. Par conséquent, pour faire des prédictions à l’échelle métagénomique, une avancée majeure dans la vitesse de prédiction est cruciale.
Meta AI a découvert que l'utilisation de modèles linguistiques de séquences protéiques accélère considérablement la prédiction de la structure, jusqu'à 60 fois. Cela suffit pour faire des prédictions sur l’intégralité d’une base de données métagénomique en quelques semaines seulement et peut être étendu à des bases de données beaucoup plus volumineuses que celles actuellement publiées. En fait, cette nouvelle capacité de prédiction de structure a permis de prédire les séquences de plus de 600 millions de protéines métagénologiques en seulement deux semaines sur un cluster d’environ 2 000 GPU.
De plus, les méthodes actuelles de prédiction de la structure SOTA nécessitent de rechercher de grandes bases de données de protéines pour identifier les séquences pertinentes. Ces méthodes nécessitent en fait un ensemble complet de séquences liées à l’évolution en entrée afin de pouvoir extraire des modèles liés à la structure. Le modèle de langage ESM-2 de Meta AI apprend ces modèles évolutifs lors de son entraînement sur les séquences protéiques, permettant des prédictions haute résolution des structures 3D directement à partir des séquences protéiques.
La figure ci-dessous montre le repliement des protéines à l'aide du modèle de langage ESM-2. Les flèches de gauche à droite montrent le flux d'informations dans le réseau depuis le modèle de langage jusqu'au coffre pliable en passant par le module structurel, et enfin les coordonnées 3D et la confiance de sortie.
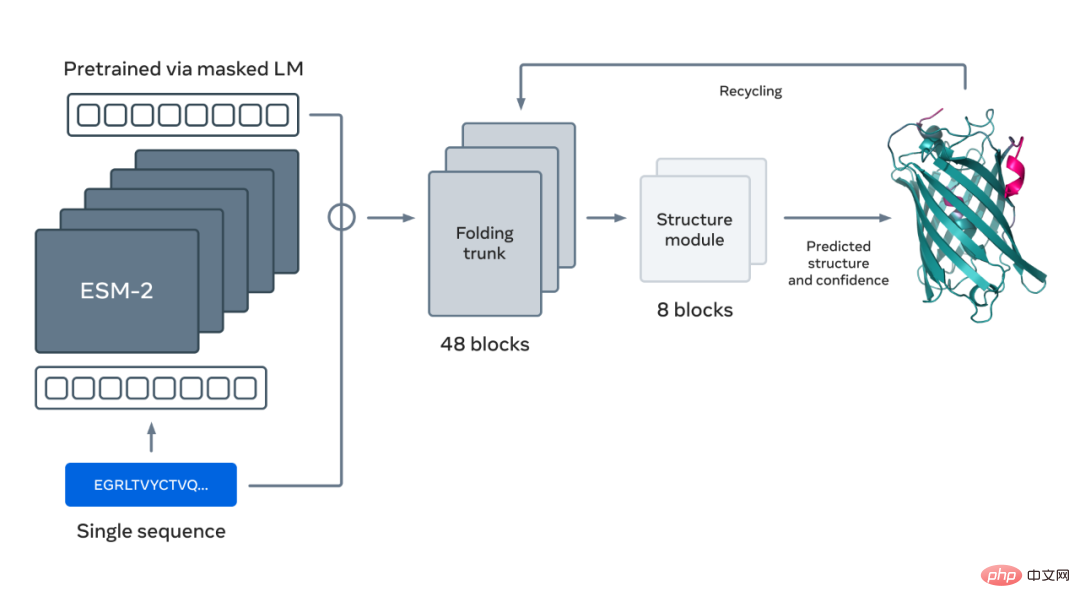
Veuillez vous référer à l'article original pour plus de détails.
Lien du blog : https://ai.facebook.com/blog/protein-folding-esmfold-metagenomics/
Ce qui précède est le contenu détaillé de. pour plus d'informations, suivez d'autres articles connexes sur le site Web de PHP en chinois!
Articles Liés
Voir plus- Tendances technologiques à surveiller en 2023
- Comment l'intelligence artificielle apporte un nouveau travail quotidien aux équipes des centres de données
- L'intelligence artificielle ou l'automatisation peuvent-elles résoudre le problème de la faible efficacité énergétique des bâtiments ?
- Co-fondateur d'OpenAI interviewé par Huang Renxun : les capacités de raisonnement de GPT-4 n'ont pas encore atteint les attentes
- Bing de Microsoft surpasse Google en termes de trafic de recherche grâce à la technologie OpenAI