
Maison >Périphériques technologiques >IA >Nouveau SOTA pour la prédiction de la fonction des protéines, méthodes d'IA basées sur les statistiques de l'Institut de technologie de Shanghai, d'Oxford et autres, publié dans la sous-journal Nature
Nouveau SOTA pour la prédiction de la fonction des protéines, méthodes d'IA basées sur les statistiques de l'Institut de technologie de Shanghai, d'Oxford et autres, publié dans la sous-journal Nature

- PHPzoriginal
- 2024-08-22 16:45:02908parcourir

Les protéines se combinent avec d'autres molécules pour faciliter presque toutes les activités biologiques de base. Par conséquent, comprendre la fonction des protéines est essentiel pour comprendre la santé, les maladies, l’évolution et la fonction de l’organisme au niveau moléculaire.
Cependant, plus de 200 millions de protéines restent non caractérisées et les méthodes informatiques s'appuient fortement sur les informations structurelles des protéines pour prédire des annotations de qualité variable.
Récemment, une équipe de recherche de l'Université d'Oxford, de l'ETH Zurich, de l'Université des sciences et technologies de Shanghai et de l'Université normale de Pékin a conçu une méthode de réseau graphique basée sur les statistiques appelée PhiGnet pour promouvoir l'annotation fonctionnelle et l'identification des sites fonctionnels des protéines.
PhiGnet surpasse non seulement les autres méthodes en termes de performances, mais réduit également l'écart séquence-fonction même en l'absence d'informations structurelles. Les résultats démontrent que l’application de l’apprentissage profond aux données évolutives peut mettre en évidence des sites fonctionnels au niveau des résidus, fournissant ainsi un soutien précieux pour l’interprétation et l’étude des propriétés existantes et des nouvelles fonctions des protéines en biomédecine.
La recherche pertinente s'intitule « Prédiction précise de la fonction des protéines à l'aide de réseaux de graphes informés par des statistiques » et a été publiée dans « Nature Communications » le 4 août.

Comprendre la fonction des protéines est essentiel pour comprendre les mécanismes complexes de nombreuses activités biologiques clés et est important pour la médecine, la biotechnologie et produits pharmaceutiques Le domaine du développement a des implications considérables.
À ce jour, plus de 356 millions de protéines ont été séquencées dans la base de données UniProt (6/2023), dont la grande majorité (~80 %) n'ont aucune annotation fonctionnelle connue.
Les méthodes d'apprentissage profond ont atteint une précision remarquable dans la prédiction des structures 3D des protéines, dépassant les capacités des méthodes classiques telles que les méthodes ab initio et la modélisation d'homologie. Cependant, attribuer avec précision des annotations fonctionnelles aux protéines reste un défi, notamment par rapport aux tests expérimentaux.
Pour relever ces défis, les chercheurs ont émis l'hypothèse que les informations contenues dans les résidus coévoluants pourraient être utilisées pour annoter les fonctions au niveau des résidus.
L'équipe de l'Université d'Oxford propose d'utiliser des réseaux de graphes basés sur des statistiques pour prédire les fonctions des protéines uniquement à partir de leurs séquences. Cette méthode caractérise intrinsèquement les caractéristiques évolutives et permet une évaluation quantitative de l’importance des résidus remplissant des fonctions spécifiques.
Cette méthode exploite les connaissances acquises à partir des données évolutives pour piloter deux réseaux convolutionnels à graphes empilés. Grâce aux connaissances acquises et à l’architecture de réseau conçue, les protéines peuvent se voir attribuer avec précision des annotations fonctionnelles et, surtout, l’importance de chaque résidu par rapport à une fonction spécifique peut être quantifiée.
PhiGnet pour l'annotation fonctionnelle des protéines
La méthode PhiGnet utilise des réseaux de graphiques basés sur des statistiques pour annoter les fonctions des protéines et identifier les sites fonctionnels des espèces en fonction de leurs séquences.

Pour absorber les connaissances du couplage évolutif (EVC) et de la communauté des résidus (RC), les chercheurs ont conçu une approche d'architecture à double canal utilisant des réseaux convolutifs à graphes empilés (GCN). Cette méthode est spécifiquement conçue pour attribuer des annotations fonctionnelles aux protéines, y compris les numéros du comité enzymatique (EC) et les termes d’ontologie des gènes (GO) (processus biologique, BP, composant cellulaire, CC et fonction moléculaire, MF).
Lorsqu'une séquence protéique est fournie, l'étude dérive son intégration à l'aide du modèle ESM-1b pré-entraîné. Par la suite, les intégrations sont entrées dans six couches convolutives de graphe d'un GCN à double pile en tant que nœuds de graphe ainsi que EVC et RC (bords de graphe). Ces couches fonctionnent en conjonction avec deux blocs de couches entièrement connectés (FC) pour traiter soigneusement les informations des deux GCN, produisant finalement un tenseur de probabilité qui évalue la faisabilité de l'attribution d'annotations fonctionnelles aux protéines.
De plus, le score d'activation dérivé à l'aide de la méthode de la carte d'activation de classe pondérée par gradient (Grad-CAM) est utilisé pour évaluer l'importance de chaque résidu dans une fonction spécifique. Ce score permet à PhiGnet d'identifier les sites fonctionnels au niveau des résidus individuels.
Par exemple, en calculant le RC de la protéine D (SdrD) contenant des répétitions sérine-aspartate, il a été démontré que les résidus des sites fonctionnels sont retenus au cours de l'évolution naturelle, et PhiGnet est capable de capturer ces informations, améliorant ainsi l'analyse de résidus. Méthodes pour prédire la fonction des protéines à un niveau de base, même en l’absence de données structurelles.
Annoter les sites fonctionnels des protéines
計算預測是否與實驗確定的功能註釋一樣準確?為了解決這個問題,研究使用活化分數對每種胺基酸對蛋白質功能的貢獻進行了定量檢查。評估了 PhiGnet 的預測性能,並評估了九種蛋白質中殘基的重要性(它們對蛋白質功能的貢獻)。
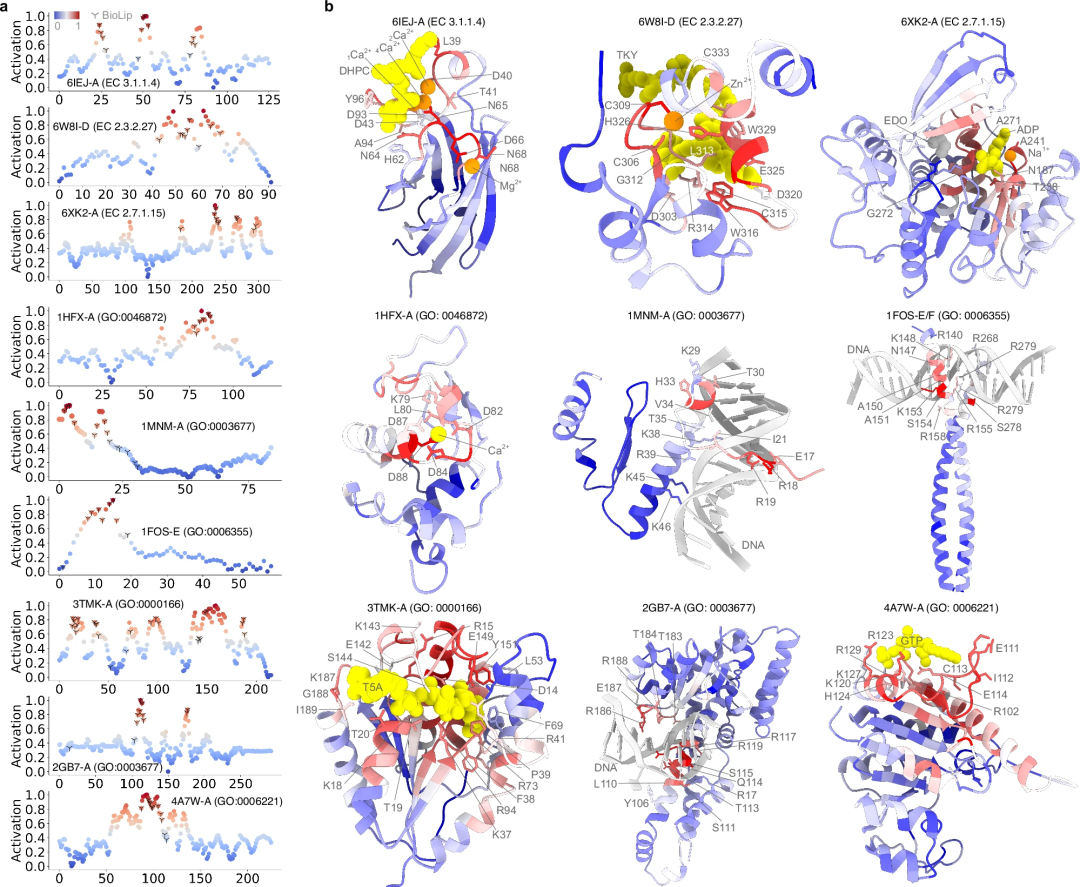
- 透過計算九種蛋白質中每個殘基的活化分數,並將它們與實驗或半手動註釋所確定的殘基進行比較。 PhiGnet 在預測殘基水平的重要位點方面表現出了良好的準確性(平均 ⩾ 75%),與實際的配體/離子/DNA 結合位點非常一致。 PhiGnet 準確地識別出具有高活化分數的蛋白質的功能重要殘基。
優於其他最先進的方法
- 為了評估 PhiGnet 的預測性能,應用該方法來推斷兩個基準測試集中蛋白質的功能註釋(EC 編號和 GO 術語)。將 PhiGnet 與最先進的方法進行比較,包括基於比對的方法、基於深度學習的方法。比較使用了兩個基本指標,包括以蛋白質為中心的 Fmax 得分和精確召回曲線下面積 (AUPR)。
圖示:不同方法在不同本體和 EC 編號中的 GO 術語之間的比較。 (資料來源:論文)
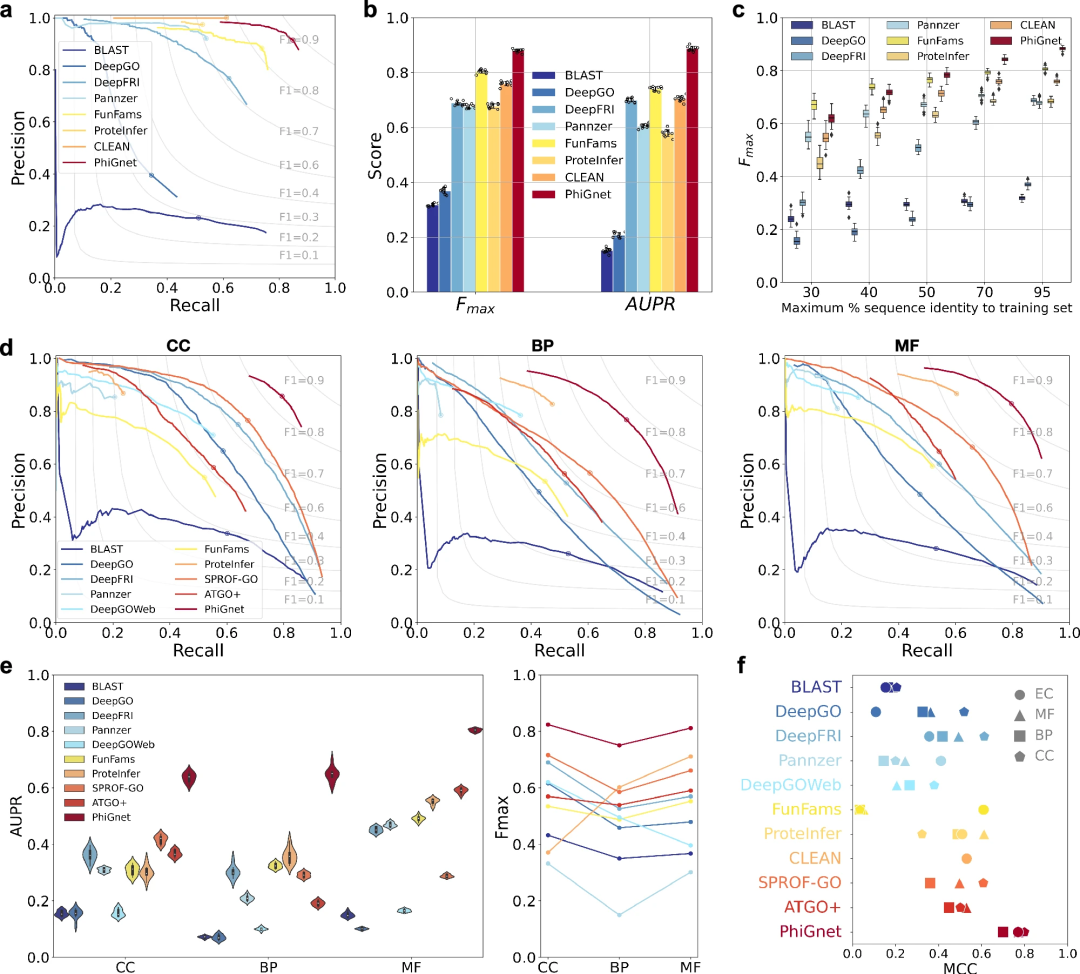
PhiGnet 展示了在兩個測試集中為蛋白質分配功能註釋的預測能力。它分別對 GO 術語和 EC 編號實現了 0.70 和 0.89 的平均 AUPR,以及 0.80 和 0.88 的 Fmax 分數。
整體而言,PhiGnet 在基準資料集上的表現明顯優於所有監督和無監督方法。
此外,還證明了 PhiGnet 的泛化穩健性,可以測試與訓練集中的蛋白質具有不同序列同一性閾值的蛋白質。在不同的最大序列同一性水平(30%、40%、50%、70% 和 95%)下,隨著序列同一性的增加,PhiGnet 表現出更好的預測性能。
由進化特徵驅動
進化數據在 PhiGnet 中起著重要作用,可用於預測蛋白質功能註釋和識別功能位點。首先,進行了消融實驗,以測試 EVC/RC 對 PhiGnet 的貢獻。實驗表明,PhiGnet 可以準確分配蛋白質功能註釋。此外,使用 EVC 或 RC 的 PhiGnet 證明了學習一般序列功能關係的強大能力,通常比其他方法更好或一樣好。
其次,進一步研究了 PhiGnet 從殘基群落中已識別的功能相關殘基中表徵有意義特徵的能力。計算了殘基的活化分數以強調它們對蛋白質功能的貢獻。值得注意的是,預測的殘基與實驗測定確定的功能位點的殘基一致,比 RC 中的殘基識別得更好。
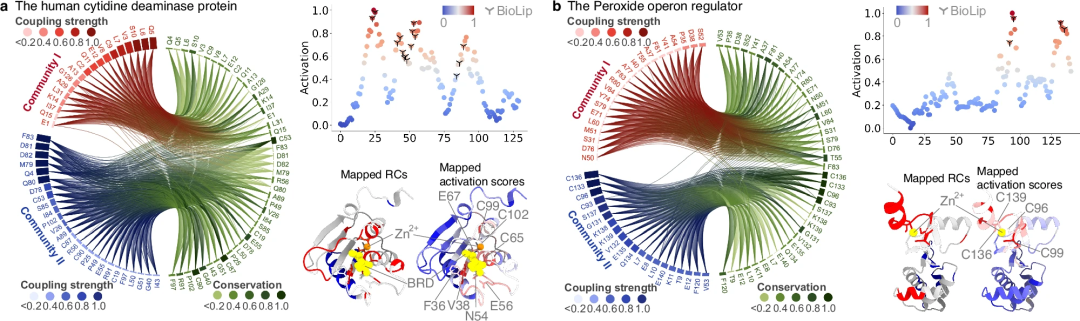
研究表明,進化信息,特別是 Remote Homology 中包含的信息,足以指定蛋白質的功能並定量表徵功能位點的殘基。此外,與 Evolutionary Vector 中較低階水平的資訊相比,Remote Homology 包含更高階水平的演化知識。同時,Remote Homology 中包含的資訊對於增強 PhiGnet 在殘留水平上識別功能相關位點的能力起著重要作用。
成功之處與局限
總之,PhiGnet 的更好性能可以歸因於它利用了蛋白質序列的進化數據和數據的高階模式,從而可以更深入、更準確地理解蛋白質功能。
PhiGnet 的主要成功之處在於利用統計資訊圖卷積神經網絡,來促進對來自海量序列資料集的演化資料的分層學習。這種方法大大超越了現有的監督和無監督方法,可用於指導未來的生物和臨床實驗。
PhiGnet 方法的限制包括序列多樣性較低的蛋白質家族中出現的偏差/噪音。將(共同)演化資訊納入 PhiGnet 可能會影響殘基群落的準確識別,特別是如果資訊來自高度保守的蛋白質家族。雖然將物理提取的知識整合到 PhiGnet 中與其他方法相比取得了顯著的改進,但在解釋 PhiGnet 中的學習機制方面仍然存在重大挑戰。
進化數據和機器學習之間的協同作用將為準確確定和設計蛋白質的生物物理特性鋪平道路。
Ce qui précède est le contenu détaillé de. pour plus d'informations, suivez d'autres articles connexes sur le site Web de PHP en chinois!
Articles Liés
Voir plus- Tendances technologiques à surveiller en 2023
- Comment l'intelligence artificielle apporte un nouveau travail quotidien aux équipes des centres de données
- L'intelligence artificielle ou l'automatisation peuvent-elles résoudre le problème de la faible efficacité énergétique des bâtiments ?
- Co-fondateur d'OpenAI interviewé par Huang Renxun : les capacités de raisonnement de GPT-4 n'ont pas encore atteint les attentes
- Bing de Microsoft surpasse Google en termes de trafic de recherche grâce à la technologie OpenAI