
Maison >Périphériques technologiques >IA >Publié dans la revue Nature, le modèle topologique Transformer prédit les interactions protéine-ligand à plusieurs échelles pour faciliter le développement de médicaments.
Publié dans la revue Nature, le modèle topologique Transformer prédit les interactions protéine-ligand à plusieurs échelles pour faciliter le développement de médicaments.

- 王林original
- 2024-07-02 15:23:211266parcourir

Une nouvelle application d'intelligence artificielle aidera les chercheurs à améliorer leurs capacités de développement de médicaments.
Le projet s'appelle TopoFormer et a été développé par une équipe interdisciplinaire dirigée par le professeur Guowei Wei du département de mathématiques de la Michigan State University.
TopoFormer transforme les informations tridimensionnelles d'une molécule en données pouvant être utilisées par des modèles d'interaction médicamenteuse typiques basés sur l'IA, étendant ainsi la capacité de ces modèles à prédire l'efficacité des médicaments.
« Grâce à l'intelligence artificielle, vous pouvez rendre le développement de médicaments plus rapide, plus efficace et moins cher », a déclaré Wei, qui est également membre du corps professoral du Département de biochimie et de biologie moléculaire et du Département de génie électrique et informatique.
Le professeur Wei a expliqué qu'aux États-Unis, le développement d'un médicament prend environ dix ans et coûte environ 2 milliards de dollars. Les essais de médicaments occupent environ la moitié du temps, tandis que l’autre moitié est consacrée à la découverte de nouveaux traitements candidats à tester.
TopoFormer a le potentiel de raccourcir le temps de développement. De cette manière, les coûts de développement des médicaments peuvent être réduits, ce qui fera baisser les prix des médicaments pour les consommateurs en aval.
L'étude s'intitulait « Transformateur structure-séquence activé par la topologie multi-échelle pour les prédictions d'interaction protéine-ligand » et a été publiée dans « Nature Machine Intelligence » le 24 juin 2024.

"Il y a plus de 20 000 protéines dans notre corps", a déclaré Wei. "Lorsqu'une maladie survient, certaines ou l'une d'entre elles devient une cible.
La première étape consiste donc à comprendre laquelle ou l'une de ces protéines est affectée." par la maladie. Quelles protéines. Ces protéines sont également ciblées par les chercheurs qui espèrent trouver des molécules capables de prévenir, d’atténuer ou de contrecarrer les effets de la maladie.
« Quand j'ai un objectif, j'essaie de trouver un grand nombre de médicaments potentiels pour cet objectif spécifique », a déclaré Wei.
Une fois que les scientifiques savent quelles protéines un médicament doit cibler, ils peuvent introduire la protéine et la séquence moléculaire du médicament potentiel dans un modèle informatique traditionnel. Ces modèles peuvent prédire la manière dont les médicaments et les cibles interagiront, guidant ainsi le développement et les médicaments à tester dans les essais cliniques.
Bien que ces modèles puissent prédire certaines interactions uniquement sur la base de la composition chimique du médicament et de la protéine, ils ignorent également les interactions importantes qui proviennent de la forme et de la structure tridimensionnelle ou 3D de la molécule.
L'ibuprofène, découvert par les chimistes dans les années 1960, en est un exemple. Il existe deux molécules d’ibuprofène différentes qui ont la même séquence chimique mais des structures 3D légèrement différentes. Un seul arrangement peut se lier aux protéines liées à la douleur et éliminer les maux de tête.
Guowei Wei a déclaré : « Les modèles d'apprentissage profond actuels ne peuvent pas expliquer la forme des médicaments ou des protéines lorsqu'ils prédisent leur interaction. »
L'architecture Transformer introduit une nouvelle technologie qui exploite le mécanisme d'attention pour l'analyse séquentielle des données entre domaines. Inspirée par cela, l'équipe de Wei a développé un modèle de transformateur topologique, TopoFormer, intégrant le laplacien hyperdigraphe topologique persistant (PTHL) au cadre Transformer.
Contrairement aux transformateurs traditionnels qui gèrent les séquences de protéines et de ligands, TopoFormer introduit des complexes protéine-ligand 3D. Il convertit ces complexes en séquences d'invariants topologiques et de formes homotopiques via PTHL, capturant ainsi leurs interactions physiques, chimiques et biologiques à plusieurs échelles.
Pré-entraîné sur divers ensembles de données, TopoFormer est capable de comprendre des interactions moléculaires complexes, y compris des effets stéréochimiques qui ne sont pas apparents dans la séquence moléculaire. Le réglage fin d'un ensemble de données spécifique peut capturer des interactions détaillées au sein d'un complexe et leurs caractéristiques par rapport à l'ensemble de données, améliorant ainsi les applications d'apprentissage profond en aval.
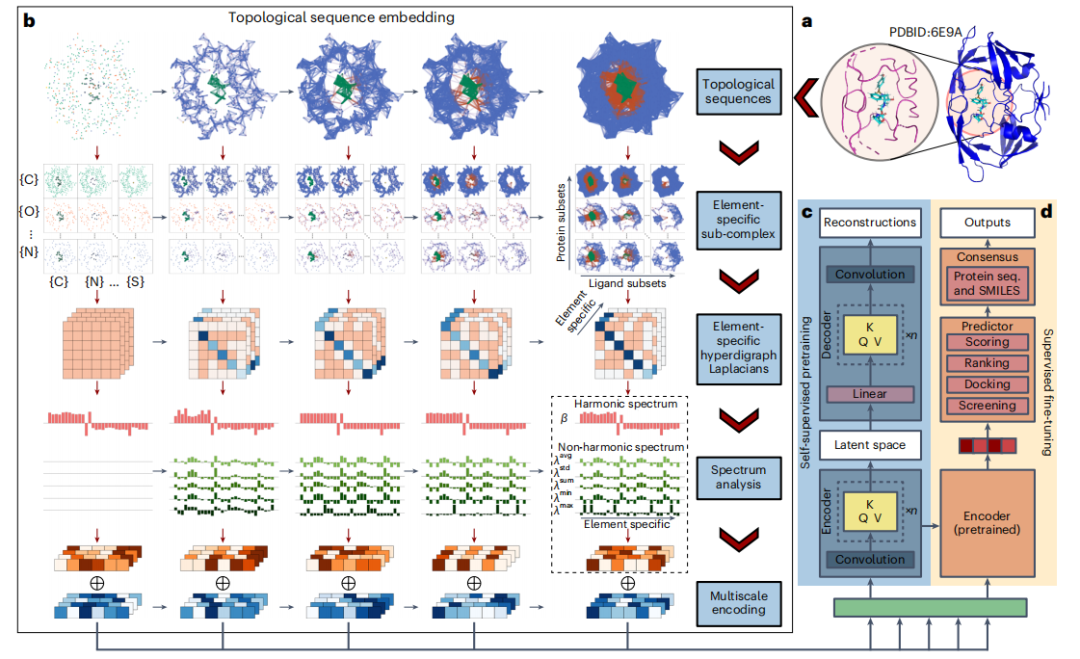
- Pour concentrer l'analyse, les chercheurs utilisent un seuil de 20 Å ou, plus précisément, de 12 Å pour identifier les réassortants et les atomes de protéines proches dans une distance définie.
- Ensuite, TopoFormer convertit la structure moléculaire 3D en une séquence topologique via le module d'intégration de séquence topologique, en utilisant PTHL pour une analyse multi-échelle. Ce processus intègre diverses interactions physiques, chimiques et biologiques dans la séquence du vecteur.
- TopoFormer utilise des complexes protéine-ligand non marqués pour un pré-entraînement auto-supervisé et un codeur-décodeur Transformer pour reconstruire des séquences topologiques. Cette étape prépare le modèle pour comprendre la dynamique protéine-ligand en l'absence de données étiquetées en mesurant la précision en comparant les intégrations de sortie et d'entrée.
- Après la pré-formation, TopoFormer entre dans la phase de réglage fin supervisé des complexes étiquetés, où les vecteurs d'intégration initiaux deviennent des éléments clés pour les tâches en aval telles que la notation, le classement, l'amarrage et le criblage. Chaque tâche possède un en-tête dédié dans le module de prédicteur.
- Pour garantir la précision et réduire les biais, TopoFormer intègre plusieurs modèles d'apprentissage profond de transformation topologique initialisés avec différentes graines et les complète avec des modèles basés sur des séquences.
- Le résultat final est un consensus de ces différentes prédictions, faisant de TopoFormer un modèle complet pour analyser les interactions protéine-ligand, exploitant à la fois les informations topologiques et l'apprentissage en profondeur.
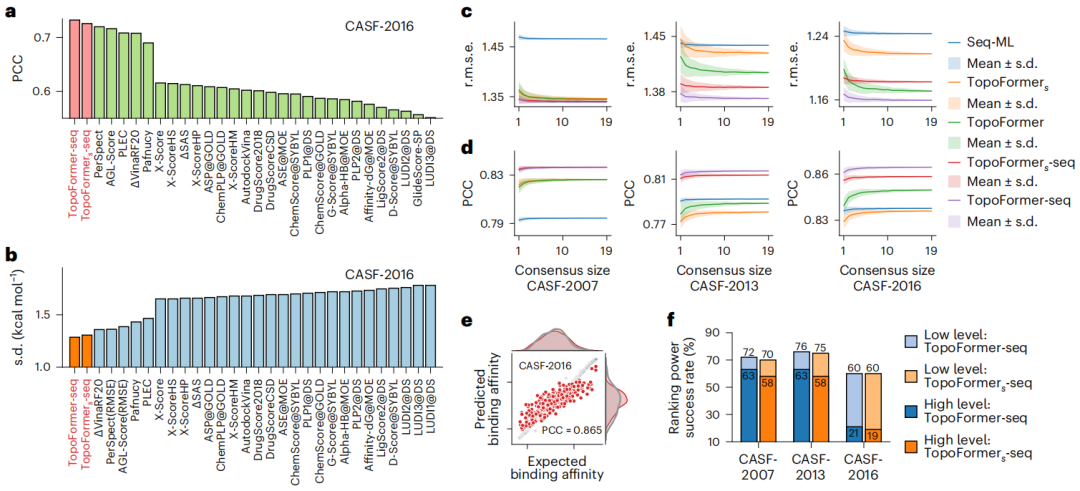
Illustration : performances de TopoFormer dans les tâches de notation et de classement. (Source : Article)
Cette approche permet d'utiliser des hyperedges dirigés de différentes dimensions pour modéliser des interactions complexes au-delà de simples connexions par paires. De plus, l’orientation de ces bords combine des propriétés physiques et chimiques, telles que l’électronégativité et l’énergie d’ionisation, pour fournir une représentation plus nuancée que les méthodes traditionnelles. Les chercheurs ont démontré cette capacité en distinguant deux isomères B7C2H9 avec des hyperbords dirigés, démontrant ainsi la capacité de la méthode à différencier efficacement les configurations élémentaires.
Lors de l'étude des complexes protéine-ligand, les chercheurs adoptent des graphiques hyperorientés topologiques comme représentations initiales et les améliorent davantage avec la théorie PTHL pour analyser leurs caractéristiques géométriques et topologiques.
S'inspirant de systèmes physiques tels que les structures moléculaires, où l'opérateur Hoch Laplacien de dimension zéro est lié à l'opérateur d'énergie cinétique de l'hamiltonien de systèmes quantiques bien définis, les chercheurs étendent l'analogie discrète aux superstructures topologiques jusqu'au graphe. Ces valeurs propres de la matrice laplacienne donnent un aperçu des propriétés des objets topologiques, analogues au spectre énergétique d'un système physique.
Par rapport à l'homologie persistante traditionnelle, la méthode PTHL marque une avancée majeure en analysant une gamme plus large de structures au-delà des complexes simplexes. Il capture des informations fondamentales sur l'homologie et des informations géométriques, notamment les nombres de Betti et l'évolution des formes homotopiques, à travers le spectre anharmonique de l'opérateur laplacien persistant.
Les résultats de l'analyse montrent qu'elle fournit une caractérisation plus complète par rapport à l'homologie traditionnelle. La multiplicité des valeurs propres nulles de l'opérateur laplacien (correspondant au nombre de Betti) confirme que cette méthode contient des informations de code-barres, fournissant un cadre puissant pour comprendre les complexes protéine-ligand.
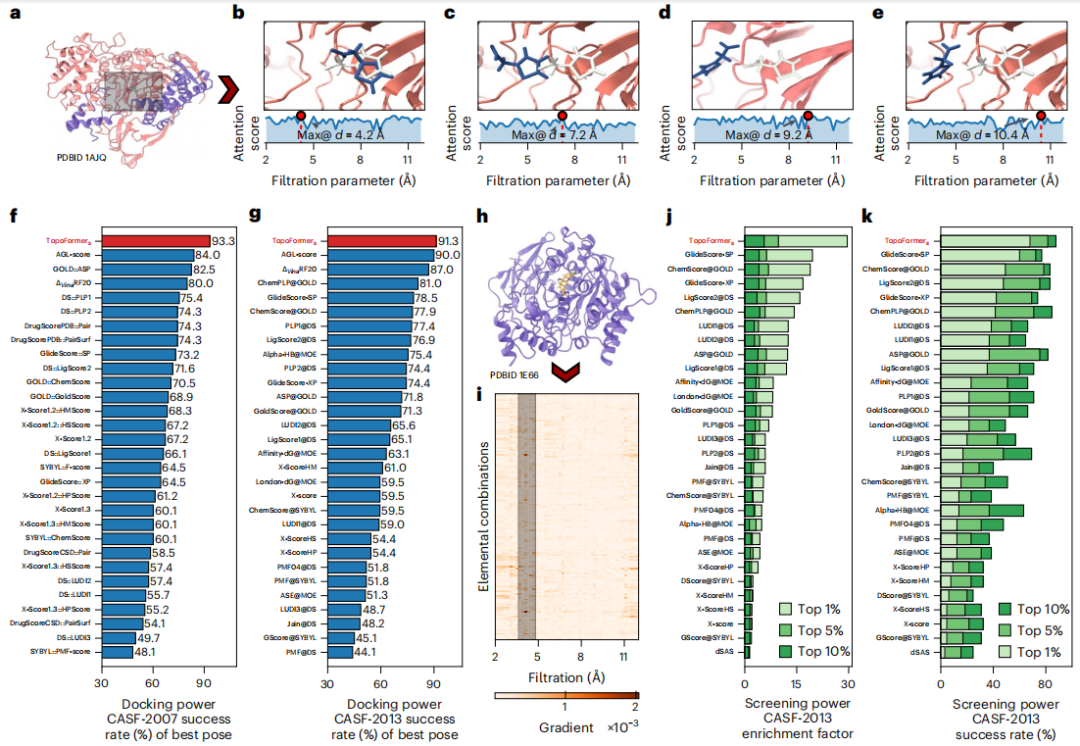
Pour capturer les interactions atomiques complexes dans les complexes protéine-ligand, y compris les forces covalentes, ioniques et de van der Waals, les chercheurs ont utilisé le PTHL pour effectuer une analyse multi-échelle. Cette approche permet d'examiner les interactions entre échelles en faisant évoluer des séquences topologiques basées sur des paramètres de filtrage, aidant ainsi le modèle Transformer à identifier le poids que chaque échelle accorde à des propriétés telles que l'affinité de liaison.
Les interactions élémentaires, notamment les liaisons hydrogène, les forces de Van der Waals et l'empilement π, constituent la base de la stabilité et de la spécificité des complexes protéine-ligand. Pour analyser ces interactions au niveau élémentaire, les chercheurs ont introduit une analyse spécifique aux éléments dans l’intégration de séquences topologiques.
Cette méthode construit des sous-hypergraphes basés sur des éléments lourds communs dans les protéines et les ligands, générant des matrices laplaciennes spécifiques aux éléments pour coder les interactions au sein du complexe. La technologie extrait des caractéristiques physiques et chimiques détaillées qui améliorent la compréhension des modèles Transformer de la dynamique complexe des interactions protéine-ligand.
Conclusion
Pour résumer, TopoFormer est formé pour lire une forme d'information et la convertir en une autre. Dans ce cas, il prend des informations tridimensionnelles sur la façon dont les protéines et les médicaments interagissent en fonction de leurs formes et les reconstruit en informations unidimensionnelles que les modèles actuels peuvent comprendre.
Le nouveau modèle est formé sur des dizaines de milliers d'interactions protéine-médicament, où chaque interaction entre deux molécules est enregistrée sous la forme d'un morceau de code ou d'un « mot ». Ces mots sont enchaînés pour former une description du complexe médicament-protéine, créant ainsi un enregistrement de sa forme.
"De cette façon, vous avez beaucoup de mots enchaînés comme des phrases.", a déclaré Wei.
D’autres modèles qui prédisent de nouvelles interactions médicamenteuses peuvent ensuite lire ces phrases et leur fournir plus de contexte. Si un nouveau médicament était un livre, TopoFormer peut transformer une idée d'histoire en une intrigue complète, prête à être écrite.
Lien papier : https://www.nature.com/articles/s42256-024-00855-1
Rapports associés : https://phys.org/news/2024-06-drug-discovery-ai-3d -typique.html
Ce qui précède est le contenu détaillé de. pour plus d'informations, suivez d'autres articles connexes sur le site Web de PHP en chinois!