
 Technology peripheralsAIShanghai Jiao Tong University team develops data-driven active learning framework to accelerate research progress in carbon nanomaterials
Technology peripheralsAIShanghai Jiao Tong University team develops data-driven active learning framework to accelerate research progress in carbon nanomaterialsShanghai Jiao Tong University team develops data-driven active learning framework to accelerate research progress in carbon nanomaterials

Editor| .
Although the controlled synthesis of substrate-catalyzed growth into carbon nanostructures is considered a promising approach, there are still challenges in dynamic catalytic surface growth mechanisms and design strategies that require further research and development.
Recently, a research team from Shanghai Jiao Tong University and Tohoku University in Japan demonstrated the effectiveness of an active machine learning model in revealing the microscopic process of substrate catalytic growth. Through the collaborative application of molecular dynamics and Monte Carlo methods, they successfully performed a comprehensive dynamic simulation of the growth of graphene on Cu(111). To enhance the accuracy of the simulation, the research team adopted a Gaussian approximation potential. This research provides new tools and methods for in-depth understanding of the catalytic growth process.
Through this research, we derived a practical and effective method that can be used to design metal or alloy substrates to obtain the desired carbon nanostructures and explore more reaction possibilities.
The research, titled "Active machine learning model for the dynamic simulation and growth mechanisms of carbon on metal surface", was published in "Nature Communications" on January 6, 2024.
 Paper link: https://www.nature.com/articles/s41467-023-44525-z
Paper link: https://www.nature.com/articles/s41467-023-44525-z
Substrate catalytic deposition is considered one of the most promising methods to achieve controllable growth of two- or three-dimensional covalently bonded networks of carbon atoms. While growth mechanisms on ordinary surfaces have been extensively studied, knowledge of the dynamic and atomic-scale factors that control graphene mass on high-index or composite surfaces is limited. This research gap has greatly hindered the development of theory-guided design approaches for novel catalytic metal substrates in the growth of carbon nanostructures.
Experimentally finding metal or alloy catalysts presents considerable challenges due to the wide range of potential substrates and the sensitivity of the carbon nanomaterial growth process to various experimental parameters.
Therefore, there is enough room for theoretical simulations and many atomic details are easily obtained. Examples include DFT, Kinetic Monte Carlo (KMC), and Ab initio Molecular Dynamics (AIMD). However, these methods each have their limitations. Therefore, there is still an urgent need for a robust design model that can accurately describe the carbon growth mechanism on metal surfaces.
Machine learning potentials (MLP) based on artificial neural networks or kernel methods are considered to be effective methods to solve the limited accuracy and transferability of classical force fields and maintain DFT-level accuracy. Despite significant achievements in data-driven MD simulations, constructing accurate MLPs remains a difficult task. One solution to this problem is dynamic learning techniques.
To improve the efficiency and effectiveness of dynamic training of deposition processes, a well-defined selection protocol is required. On the other hand, the dynamics of carbon growth on metallic substrates can be controlled by important rare events. Therefore, how to improve the training efficiency of MLP by combining boosted sampling methods with classical dynamics requires further research.
Data-driven automatic learning framework to generate MLP with minimum manpowerThis study proposes a data-driven automatic learning framework to generate MLP with minimum manpower , which is suitable for carbon growth on metal or alloy surfaces.
To achieve this task, the researchers used (1) Gaussian approximation potential (GAP) processing learning model; (2) an enhanced sampling method called timestamp force bias Monte Carlo (time-stamped force-biased Monte Carlo, tfMC) method to accelerate the relaxation process after carbon deposition, thereby including important rare events in the training database; (3) Smooth overlap based on atomic positions (SOAP) Efficient strategies for descriptor selection of representative training data; (4) well-established carbon training sets; (5) automated screening, fitting, and validation procedures.
 Figure 1: Carbon growth on metal generated by dynamic active learning machine learning potential (CGM) during a hybrid MD/tfMC simulation -MLP) schematic diagram. (Source: Paper)
Figure 1: Carbon growth on metal generated by dynamic active learning machine learning potential (CGM) during a hybrid MD/tfMC simulation -MLP) schematic diagram. (Source: Paper)
By exploiting the high accuracy of the Carbon Growth Machine Learning Potential (CGM-MLP) and incorporating rare atomic events in the MD/tfMC method, we successfully replicated graphene with metal surfaces The basic subprocesses related to nucleation and carbon growth are shown in the figure below.

Figure 2: CGM-MLP driven simulation of graphene growth on Cu(111) with different carbon incident kinetic energies (Ek). (Source: Paper)
The resulting potential was then applied to study the deposition growth of carbon atoms on the Cu (111) surface. This method can correctly capture the key processes of carbon growth on Cu(111), such as the formation and migration of subsurface carbon monomers and surface dimers, the emergence of one-dimensional carbon nanocrystals, graphene nucleation involving Cu atoms and Edge passivation of carbon chains, and precipitation growth process.
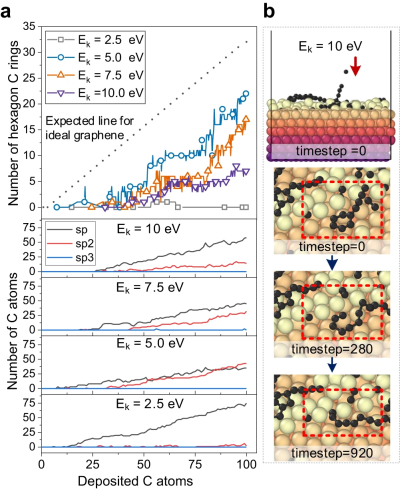
Figure 3: Carbon structure analysis and observation of carbon ring breakage by high-energy bombardment. (Source: Paper)
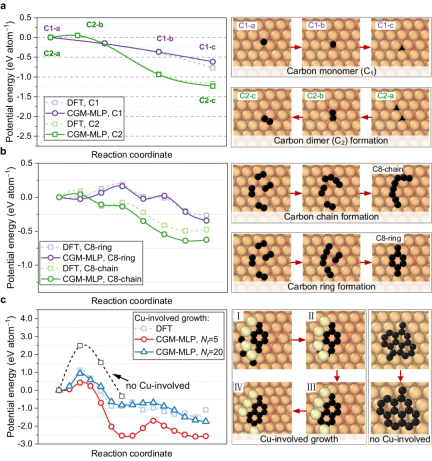
#Figure 4: Carbon diffusion and graphite obtained using CGM-MLP on metal and DFT-based crawling image nudging elastic band (CI-NEB) calculations The minimum energy path for ene nucleation. (Source: Paper)
Researchers investigated the initial nucleation of different metal surfaces, especially carbon deposition on Cu(111), Cr(110), Ti(001) and O-contaminated Cu(111) The simulations show consistency with experimental observations and DFT calculations.
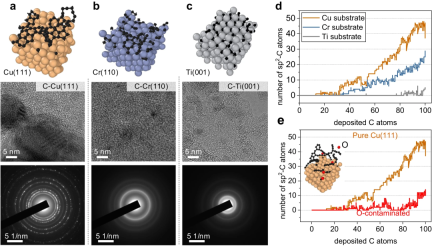
Figure 5: Representative metal surfaces for carbon nanostructure growth. (Source: Paper)
Research Significance
In summary, this research represents a pioneering advance in the integration of MLP and MD/tfMC for designing metal or alloy substrates provides a transferable and efficient strategy to obtain desired carbon nanostructures.
CGM-MLP effectively combines the accuracy of first principles methods with the efficiency of classical force fields. The tfMC method overcomes the time scale limitations of traditional AIMD or classical MD methods. Furthermore, the automated training framework of CGM-MLP incorporates specialized query strategies for building dynamic training sets in deposition simulations, emphasizing the importance of considering the local environment around deposited atoms.
These developments make it possible to directly study the mechanism of carbon growth on complex metal surfaces. The machine learning-driven deposition model proposed in this study may provide opportunities to study the growth of a variety of carbon nanostructures (e.g., graphene, carbon nanotubes, graphite, or diamond-like carbon films) on multi-element metal or alloy substrates.
The above is the detailed content of Shanghai Jiao Tong University team develops data-driven active learning framework to accelerate research progress in carbon nanomaterials. For more information, please follow other related articles on the PHP Chinese website!

 The AI Skills Gap Is Slowing Down Supply ChainsApr 26, 2025 am 11:13 AM
The AI Skills Gap Is Slowing Down Supply ChainsApr 26, 2025 am 11:13 AMThe term "AI-ready workforce" is frequently used, but what does it truly mean in the supply chain industry? According to Abe Eshkenazi, CEO of the Association for Supply Chain Management (ASCM), it signifies professionals capable of critic
 How One Company Is Quietly Working To Transform AI ForeverApr 26, 2025 am 11:12 AM
How One Company Is Quietly Working To Transform AI ForeverApr 26, 2025 am 11:12 AMThe decentralized AI revolution is quietly gaining momentum. This Friday in Austin, Texas, the Bittensor Endgame Summit marks a pivotal moment, transitioning decentralized AI (DeAI) from theory to practical application. Unlike the glitzy commercial
 Nvidia Releases NeMo Microservices To Streamline AI Agent DevelopmentApr 26, 2025 am 11:11 AM
Nvidia Releases NeMo Microservices To Streamline AI Agent DevelopmentApr 26, 2025 am 11:11 AMEnterprise AI faces data integration challenges The application of enterprise AI faces a major challenge: building systems that can maintain accuracy and practicality by continuously learning business data. NeMo microservices solve this problem by creating what Nvidia describes as "data flywheel", allowing AI systems to remain relevant through continuous exposure to enterprise information and user interaction. This newly launched toolkit contains five key microservices: NeMo Customizer handles fine-tuning of large language models with higher training throughput. NeMo Evaluator provides simplified evaluation of AI models for custom benchmarks. NeMo Guardrails implements security controls to maintain compliance and appropriateness
 AI Paints A New Picture For The Future Of Art And DesignApr 26, 2025 am 11:10 AM
AI Paints A New Picture For The Future Of Art And DesignApr 26, 2025 am 11:10 AMAI: The Future of Art and Design Artificial intelligence (AI) is changing the field of art and design in unprecedented ways, and its impact is no longer limited to amateurs, but more profoundly affecting professionals. Artwork and design schemes generated by AI are rapidly replacing traditional material images and designers in many transactional design activities such as advertising, social media image generation and web design. However, professional artists and designers also find the practical value of AI. They use AI as an auxiliary tool to explore new aesthetic possibilities, blend different styles, and create novel visual effects. AI helps artists and designers automate repetitive tasks, propose different design elements and provide creative input. AI supports style transfer, which is to apply a style of image
 How Zoom Is Revolutionizing Work With Agentic AI: From Meetings To MilestonesApr 26, 2025 am 11:09 AM
How Zoom Is Revolutionizing Work With Agentic AI: From Meetings To MilestonesApr 26, 2025 am 11:09 AMZoom, initially known for its video conferencing platform, is leading a workplace revolution with its innovative use of agentic AI. A recent conversation with Zoom's CTO, XD Huang, revealed the company's ambitious vision. Defining Agentic AI Huang d
 The Existential Threat To UniversitiesApr 26, 2025 am 11:08 AM
The Existential Threat To UniversitiesApr 26, 2025 am 11:08 AMWill AI revolutionize education? This question is prompting serious reflection among educators and stakeholders. The integration of AI into education presents both opportunities and challenges. As Matthew Lynch of The Tech Edvocate notes, universit
 The Prototype: American Scientists Are Looking For Jobs AbroadApr 26, 2025 am 11:07 AM
The Prototype: American Scientists Are Looking For Jobs AbroadApr 26, 2025 am 11:07 AMThe development of scientific research and technology in the United States may face challenges, perhaps due to budget cuts. According to Nature, the number of American scientists applying for overseas jobs increased by 32% from January to March 2025 compared with the same period in 2024. A previous poll showed that 75% of the researchers surveyed were considering searching for jobs in Europe and Canada. Hundreds of NIH and NSF grants have been terminated in the past few months, with NIH’s new grants down by about $2.3 billion this year, a drop of nearly one-third. The leaked budget proposal shows that the Trump administration is considering sharply cutting budgets for scientific institutions, with a possible reduction of up to 50%. The turmoil in the field of basic research has also affected one of the major advantages of the United States: attracting overseas talents. 35
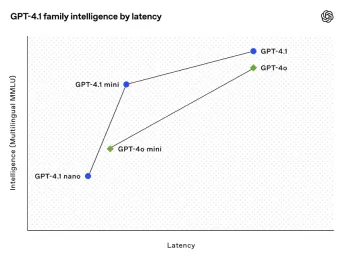 All About Open AI's Latest GPT 4.1 Family - Analytics VidhyaApr 26, 2025 am 10:19 AM
All About Open AI's Latest GPT 4.1 Family - Analytics VidhyaApr 26, 2025 am 10:19 AMOpenAI unveils the powerful GPT-4.1 series: a family of three advanced language models designed for real-world applications. This significant leap forward offers faster response times, enhanced comprehension, and drastically reduced costs compared t


Hot AI Tools

Undresser.AI Undress
AI-powered app for creating realistic nude photos

AI Clothes Remover
Online AI tool for removing clothes from photos.

Undress AI Tool
Undress images for free

Clothoff.io
AI clothes remover

Video Face Swap
Swap faces in any video effortlessly with our completely free AI face swap tool!

Hot Article
Hot Tools

WebStorm Mac version
Useful JavaScript development tools

mPDF
mPDF is a PHP library that can generate PDF files from UTF-8 encoded HTML. The original author, Ian Back, wrote mPDF to output PDF files "on the fly" from his website and handle different languages. It is slower than original scripts like HTML2FPDF and produces larger files when using Unicode fonts, but supports CSS styles etc. and has a lot of enhancements. Supports almost all languages, including RTL (Arabic and Hebrew) and CJK (Chinese, Japanese and Korean). Supports nested block-level elements (such as P, DIV),

EditPlus Chinese cracked version
Small size, syntax highlighting, does not support code prompt function

DVWA
Damn Vulnerable Web App (DVWA) is a PHP/MySQL web application that is very vulnerable. Its main goals are to be an aid for security professionals to test their skills and tools in a legal environment, to help web developers better understand the process of securing web applications, and to help teachers/students teach/learn in a classroom environment Web application security. The goal of DVWA is to practice some of the most common web vulnerabilities through a simple and straightforward interface, with varying degrees of difficulty. Please note that this software

SublimeText3 English version
Recommended: Win version, supports code prompts!

