

 Technology peripheralsAIPeking University & Wangshi Intelligence proposes a new model: bridging the gap between chemical reaction pre-training and conditional molecule generation!
Technology peripheralsAIPeking University & Wangshi Intelligence proposes a new model: bridging the gap between chemical reaction pre-training and conditional molecule generation!
Chemical reactions are the basis of drug design and organic chemistry research. There is a growing need among the research community for a large-scale deep learning framework that can effectively capture the fundamental rules of chemical reactions.
Recently, a research team from Peking University and Wangshi Intelligence proposed a new method to bridge the gap between reaction-based molecular pre-training and generation tasks.
Inspired by the mechanisms of organic chemistry, researchers have developed a new pre-training framework that enables it to incorporate inductive bias into models. This proposed framework achieves state-of-the-art results when performing challenging downstream tasks. By leveraging knowledge of chemistry, the framework overcomes the limitations of current molecular generation models that rely on a small number of reaction templates. Across extensive experiments, the model generated high-quality, synthesizable drug-like structures
Overall, this research leads to large-scale deep learning for a variety of reaction-based applications The framework has taken an important step forward.
The research was titled "Bridging the gap between chemical reaction pretraining and conditional molecule generation with a unified model" and was published in "Nature Machine Intelligence" on December 5, 2023 .

Paper link: https://www.nature.com/articles/s42256-023-00764-9
Deep learning models have been widely used in many scientific research fields. The pre-training framework plays a positive role in the seamless integration of new tasks and can speed up the modeling process, especially when labeled data is limited
The basis of drug design and organic chemistry research It's a chemical reaction. Currently, the research and application of data mining have enabled deep learning models to be used in chemical reactions. Based on these data, there have been many data-driven studies that delve into representation learning of chemical reactions
Representation learning refers to automatically learning useful features from data and then using them for various Downstream tasks. Existing methods ignore basic theories of organic chemistry, limiting their performance.
Molecular generation based on chemical reactions
In addition to the reaction classification task, molecule generation based on chemical reactions is also an important application. In earlier studies, template-based stepwise molecule generation strategies were often employed
These template-based methods relied heavily on predefined building blocks and reactions, which narrowed the Accessible chemical space. A similar trend is found in the field of reaction product prediction, where template-based methods cannot be extrapolated to complex reactions; this problem can be solved by using template-free methods.
In reaction-based molecule generation tasks, template-free methods also show generalization advantages over template-based methods. However, existing template-free molecule generation methods can only generate molecules based on predefined reactant libraries. In addition to this, for the lead compound or lead optimization stage in drug design, it is more advantageous to utilize chemical reactions as editing tools to modify a given structure. The resulting chemical library will focus on a subset of chemical space that can be synthesized with fewer reaction steps.
A new, comprehensive deep learning framework for chemical reactions
Here, researchers propose a new, comprehensive deep learning framework for chemical reactions called Uni -RXN. It aims to solve two basic tasks: self-supervised representation learning and conditional generative modeling.
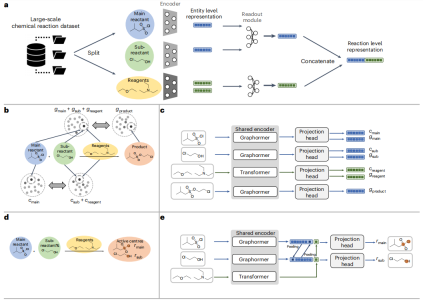
Show: Uni-RXN composition and method. (Quote from: paper)
Different from existing methods, the researchers proposed a set of self-supervised tasks specifically designed for chemical reactions. These tasks include reaction center prediction, primary reactant and subreactant pairing, and reactant-product pairing. In an extensive evaluation on challenging reaction tasks, the Uni-RXN method surpasses the state-of-the-art, demonstrating its ability to effectively capture domain knowledge of chemical reactions. The promising results obtained pave the way for widespread downstream applications
By effectively capturing chemical rules, Uni-RXN is well suited for generation tasks. Unlike traditional methods that rely on selecting fragments from a library of predefined reactants, Uni-RXN takes molecular structures as input conditions and generates representations of the corresponding reactants while maintaining permutation invariance within the reaction. Leveraging the power of the dense vector similarity search package, Uni-RXN enables efficient retrieval of reactants from large reactant and reagent libraries. Subsequently, a reaction prediction model is employed to generate product outputs.
Compared to template-based methods that explore only a limited subset of chemical space, Uni-RXN exhibits superior performance in generating a wider range of synthesizable drug-like structures. This feature makes it particularly suitable for virtual library enumeration, and is supported by comprehensive statistical analysis and case studies.
The Uni-RXN approach has many advantages and can generate rich representations for the challenging chemical reaction classification task. Compared with other baseline models, Uni-RXN achieves an accuracy of 58.7% with only 4 data points per category
Rewritten content: Chemical reaction classification See Table 1 for accuracy. (Source: Paper)
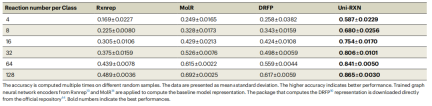
Transformer models can be used to differentiate between optimized and unoptimized chemical reaction data. In addition, the encoder can also be easily applied to the generation of structural conditions
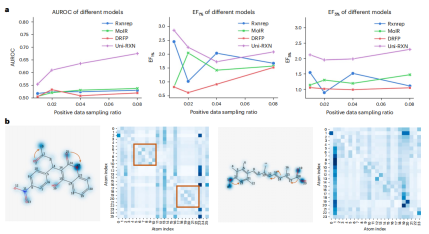
The content that needs to be rewritten is: The chart shows the retrieval performance of Uni-RXN and attention weight. (Source: Paper)
The results highlight the favorable properties of molecules generated by the proposed model, which make them well suited for drug discovery tasks. The model can generate more molecules with drug-like properties and synthesizability
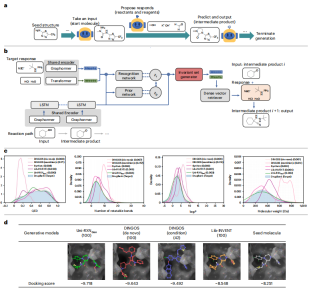
Illustration: Uni-RXNGen process and performance. (Source: paper)
Combined with virtual screening methods such as molecular docking, this generated model can achieve efficient structure-activity relationship research. The huge synthetic drug-like chemical space generated by this model can improve the true positive rate of drug repurposing or hit molecule searches.
The above is the detailed content of Peking University & Wangshi Intelligence proposes a new model: bridging the gap between chemical reaction pre-training and conditional molecule generation!. For more information, please follow other related articles on the PHP Chinese website!

 You Must Build Workplace AI Behind A Veil Of IgnoranceApr 29, 2025 am 11:15 AM
You Must Build Workplace AI Behind A Veil Of IgnoranceApr 29, 2025 am 11:15 AMIn John Rawls' seminal 1971 book The Theory of Justice, he proposed a thought experiment that we should take as the core of today's AI design and use decision-making: the veil of ignorance. This philosophy provides a simple tool for understanding equity and also provides a blueprint for leaders to use this understanding to design and implement AI equitably. Imagine that you are making rules for a new society. But there is a premise: you don’t know in advance what role you will play in this society. You may end up being rich or poor, healthy or disabled, belonging to a majority or marginal minority. Operating under this "veil of ignorance" prevents rule makers from making decisions that benefit themselves. On the contrary, people will be more motivated to formulate public
 Decisions, Decisions… Next Steps For Practical Applied AIApr 29, 2025 am 11:14 AM
Decisions, Decisions… Next Steps For Practical Applied AIApr 29, 2025 am 11:14 AMNumerous companies specialize in robotic process automation (RPA), offering bots to automate repetitive tasks—UiPath, Automation Anywhere, Blue Prism, and others. Meanwhile, process mining, orchestration, and intelligent document processing speciali
 The Agents Are Coming – More On What We Will Do Next To AI PartnersApr 29, 2025 am 11:13 AM
The Agents Are Coming – More On What We Will Do Next To AI PartnersApr 29, 2025 am 11:13 AMThe future of AI is moving beyond simple word prediction and conversational simulation; AI agents are emerging, capable of independent action and task completion. This shift is already evident in tools like Anthropic's Claude. AI Agents: Research a
 Why Empathy Is More Important Than Control For Leaders In An AI-Driven FutureApr 29, 2025 am 11:12 AM
Why Empathy Is More Important Than Control For Leaders In An AI-Driven FutureApr 29, 2025 am 11:12 AMRapid technological advancements necessitate a forward-looking perspective on the future of work. What happens when AI transcends mere productivity enhancement and begins shaping our societal structures? Topher McDougal's upcoming book, Gaia Wakes:
 AI For Product Classification: Can Machines Master Tax Law?Apr 29, 2025 am 11:11 AM
AI For Product Classification: Can Machines Master Tax Law?Apr 29, 2025 am 11:11 AMProduct classification, often involving complex codes like "HS 8471.30" from systems such as the Harmonized System (HS), is crucial for international trade and domestic sales. These codes ensure correct tax application, impacting every inv
 Could Data Center Demand Spark A Climate Tech Rebound?Apr 29, 2025 am 11:10 AM
Could Data Center Demand Spark A Climate Tech Rebound?Apr 29, 2025 am 11:10 AMThe future of energy consumption in data centers and climate technology investment This article explores the surge in energy consumption in AI-driven data centers and its impact on climate change, and analyzes innovative solutions and policy recommendations to address this challenge. Challenges of energy demand: Large and ultra-large-scale data centers consume huge power, comparable to the sum of hundreds of thousands of ordinary North American families, and emerging AI ultra-large-scale centers consume dozens of times more power than this. In the first eight months of 2024, Microsoft, Meta, Google and Amazon have invested approximately US$125 billion in the construction and operation of AI data centers (JP Morgan, 2024) (Table 1). Growing energy demand is both a challenge and an opportunity. According to Canary Media, the looming electricity
 AI And Hollywood's Next Golden AgeApr 29, 2025 am 11:09 AM
AI And Hollywood's Next Golden AgeApr 29, 2025 am 11:09 AMGenerative AI is revolutionizing film and television production. Luma's Ray 2 model, as well as Runway's Gen-4, OpenAI's Sora, Google's Veo and other new models, are improving the quality of generated videos at an unprecedented speed. These models can easily create complex special effects and realistic scenes, even short video clips and camera-perceived motion effects have been achieved. While the manipulation and consistency of these tools still need to be improved, the speed of progress is amazing. Generative video is becoming an independent medium. Some models are good at animation production, while others are good at live-action images. It is worth noting that Adobe's Firefly and Moonvalley's Ma
 Is ChatGPT Slowly Becoming AI's Biggest Yes-Man?Apr 29, 2025 am 11:08 AM
Is ChatGPT Slowly Becoming AI's Biggest Yes-Man?Apr 29, 2025 am 11:08 AMChatGPT user experience declines: is it a model degradation or user expectations? Recently, a large number of ChatGPT paid users have complained about their performance degradation, which has attracted widespread attention. Users reported slower responses to models, shorter answers, lack of help, and even more hallucinations. Some users expressed dissatisfaction on social media, pointing out that ChatGPT has become “too flattering” and tends to verify user views rather than provide critical feedback. This not only affects the user experience, but also brings actual losses to corporate customers, such as reduced productivity and waste of computing resources. Evidence of performance degradation Many users have reported significant degradation in ChatGPT performance, especially in older models such as GPT-4 (which will soon be discontinued from service at the end of this month). this


Hot AI Tools

Undresser.AI Undress
AI-powered app for creating realistic nude photos

AI Clothes Remover
Online AI tool for removing clothes from photos.

Undress AI Tool
Undress images for free

Clothoff.io
AI clothes remover

Video Face Swap
Swap faces in any video effortlessly with our completely free AI face swap tool!

Hot Article
Hot Tools

SAP NetWeaver Server Adapter for Eclipse
Integrate Eclipse with SAP NetWeaver application server.

Atom editor mac version download
The most popular open source editor

SecLists
SecLists is the ultimate security tester's companion. It is a collection of various types of lists that are frequently used during security assessments, all in one place. SecLists helps make security testing more efficient and productive by conveniently providing all the lists a security tester might need. List types include usernames, passwords, URLs, fuzzing payloads, sensitive data patterns, web shells, and more. The tester can simply pull this repository onto a new test machine and he will have access to every type of list he needs.

Zend Studio 13.0.1
Powerful PHP integrated development environment

EditPlus Chinese cracked version
Small size, syntax highlighting, does not support code prompt function

