

 Technology peripheralsAIFaster than 0! Meta launched a large protein model with 15 billion parameters to crush AlphaFold2
Technology peripheralsAIFaster than 0! Meta launched a large protein model with 15 billion parameters to crush AlphaFold2Faster than 0! Meta launched a large protein model with 15 billion parameters to crush AlphaFold2

The largest protein language model to date has been released!
A year ago, DeepMind’s open source AlphaFold2 was launched in Nature and Science, overwhelming the biological and AI academic circles.
A year later, Meta came with ESMFold, which was an order of magnitude faster.
Not only is it fast, the model also has 15 billion parameters.

LeCun tweeted to praise this as a great new achievement by the Meta-FAIR protein team.

Co-author Zeming Lin revealed that the large model with 3 billion parameters was trained on 256 GPUs for 3 weeks, while ESMfold took 10 days on 128 GPUs. As for the 15 billion parameter version, it is still unclear.
He also said that the code will definitely be open sourced later, so stay tuned!
Big and fast!
Today, our protagonist is ESMFold, a model that directly predicts high-accuracy, end-to-end, atomic-level structure from individual protein sequences.

Paper address: https://www.biorxiv.org/content/10.1101/2022.07.20.500902v1
The benefits of 15 billion parameters Needless to say – today’s large models can be trained to predict the three-dimensional structure of proteins with atomic-sized accuracy.
In terms of accuracy, ESMFold is similar to AlphaFold2 and RoseTTAFold.
However, ESMFold’s inference speed is an order of magnitude faster than AlphaFold2!
It may be difficult to understand the speed comparison between the three by talking about the order of magnitude. Just look at the picture below to understand.

What’s the difference?
Although AlphaFold2 and RoseTTAFold have achieved breakthrough success in the problem of atomic resolution structure prediction, they also rely on the use of multiple sequence alignments (MSA) and similar protein structure templates for optimal performance.
In contrast, by leveraging the internal representation of the language model, ESMFold can generate corresponding structure predictions using only one sequence as input, thus greatly speeding up structure prediction.

The researchers found that ESMFold’s predictions for low-complexity sequences were comparable to current state-of-the-art models.
Moreover, the accuracy of structure prediction is closely related to the complexity of the language model. That is to say, when the language model can better understand the sequence, it can better understand the structure.

Currently, there are billions of protein sequences of unknown structure and function, many of which are derived from metagenomic sequencing.
Using ESMFold, researchers can fold a random sample of 1 million metagenomic sequences in just 6 hours.

A large proportion of these have high confidence and are unlike any known structure (have no records in the database).
Researchers believe that ESMFold can help understand protein structures that are beyond current understanding.

Additionally, because ESMFold’s predictions are an order of magnitude faster than existing models, researchers can use ESMFold to help fill rapidly growing protein sequence databases and slow progress. The gap between protein structure and function databases.
15 billion parameter protein language model
Next let’s talk about Meta’s new ESMFold in detail.
ESM-2 is a Transformer-based language model and uses an attention mechanism to learn the interaction patterns between pairs of amino acids in the input sequence.
Compared with the previous generation model ESM-1b, Meta has improved the model structure and training parameters, and added computing resources and data. At the same time, the addition of relative position embedding enables the model to be generalized to sequences of any length.
From the results, the ESM-2 model with 150 million parameters performed better than the ESM-1b model with 650 million parameters.
In addition, ESM-2 also surpasses other protein language models on the benchmark of structure prediction. This performance improvement is consistent with established patterns in the large language modeling field.

As the scale of ESM-2 increases, it can be observed that the accuracy of language modeling has greatly improved.

End-to-end single sequence structure prediction
A key difference between SMFold and AlphaFold2 is that ESMFold uses language model representation, which eliminates the need for explicit homology Sequences (in the form of MSA) are required as input.
ESMFold simplifies the Evoformer in AlphaFold2 by replacing the computationally expensive network module that handles MSA with a Transformer module that handles sequences. This simplification means that ESMFold is significantly faster than MSA-based models.
The output of the folded backbone is then processed by a structure module, which is responsible for outputting the final atomic-level structure and prediction confidence.

Researchers compared ESMFold with AlphaFold2 and RoseTTAFold on the CAMEO (April 2022 to June 2022) and CASP14 (May 2020) test sets.
When only a single sequence is given as input, ESMFold performs much better than Alphafold 2.
When using the complete pipeline, AlphaFold2 achieved 88.3 and 84.7 on CAMEO and CASP14 respectively. ESMFold achieves comparable accuracy to RoseTTAfold on CAMEO, with an average TM score of 82.0.

Conclusion
The researchers found that language models targeting unsupervised learning performed well on a large Trained on an evolutionarily diverse protein sequence database, it can predict protein structures with atomic-level resolution.
By expanding the parameters of the language model to 15B, the impact of scale on protein structure learning can be systematically studied.
We saw that the nonlinear curve of protein structure predictions is a function of model size, and observed a strong connection between how well a language model understands a sequence and its structure predictions.
The models of the ESM-2 series are the largest protein language models trained to date, with only an order of magnitude fewer parameters than the largest recently developed text models.
Moreover, ESM-2 is a very big improvement over the previous model. Even under 150M parameters, ESM-2 captures more accurately than the ESM-1 generation language model under 650 million parameters. Structure diagram.
Researchers said that the biggest driver of ESMFold performance is the language model. Because there is a strong connection between the perplexity of language models and the accuracy of structure predictions, they found that when ESM-2 can better understand protein sequences, it can achieve predictions comparable to current state-of-the-art models.
ESMFold has obtained accurate atomic resolution structure prediction, and the inference time is an order of magnitude faster than AlphaFold2.
In practice, the speed advantage is even greater. Because ESMFold does not need to search for evolutionarily related sequences to construct MSA.
Although there are faster ways to reduce search time, it may still be very long no matter how much it is reduced.
The benefits brought by the greatly shortened inference time are self-evident - the increase in speed will make it possible to map the structural space of large metagenomics sequence databases.
In addition to structure-based tools to identify distal homology and conservation, rapid and accurate structure prediction with ESMFold can also play an important role in the structural and functional analysis of large new sequence collections.
Obtaining millions of predicted structures in a limited time will help discover new understanding of the breadth and diversity of natural proteins and enable the discovery of completely new protein structures and protein functions.
Introduction to the author
The co-author of this article is Zeming Lin from Meta AI.

According to his personal homepage, Zeming studied for a PhD at New York University and worked as a research engineer (visiting) at Meta AI, mainly responsible for back-end infrastructure work.
He studied at the University of Virginia for both his bachelor's and master's degrees, where he and Yanjun Qi did research on machine learning applications, especially in protein structure prediction.
The areas of interest are deep learning, structure prediction, and information biology.
The above is the detailed content of Faster than 0! Meta launched a large protein model with 15 billion parameters to crush AlphaFold2. For more information, please follow other related articles on the PHP Chinese website!

 The AI Skills Gap Is Slowing Down Supply ChainsApr 26, 2025 am 11:13 AM
The AI Skills Gap Is Slowing Down Supply ChainsApr 26, 2025 am 11:13 AMThe term "AI-ready workforce" is frequently used, but what does it truly mean in the supply chain industry? According to Abe Eshkenazi, CEO of the Association for Supply Chain Management (ASCM), it signifies professionals capable of critic
 How One Company Is Quietly Working To Transform AI ForeverApr 26, 2025 am 11:12 AM
How One Company Is Quietly Working To Transform AI ForeverApr 26, 2025 am 11:12 AMThe decentralized AI revolution is quietly gaining momentum. This Friday in Austin, Texas, the Bittensor Endgame Summit marks a pivotal moment, transitioning decentralized AI (DeAI) from theory to practical application. Unlike the glitzy commercial
 Nvidia Releases NeMo Microservices To Streamline AI Agent DevelopmentApr 26, 2025 am 11:11 AM
Nvidia Releases NeMo Microservices To Streamline AI Agent DevelopmentApr 26, 2025 am 11:11 AMEnterprise AI faces data integration challenges The application of enterprise AI faces a major challenge: building systems that can maintain accuracy and practicality by continuously learning business data. NeMo microservices solve this problem by creating what Nvidia describes as "data flywheel", allowing AI systems to remain relevant through continuous exposure to enterprise information and user interaction. This newly launched toolkit contains five key microservices: NeMo Customizer handles fine-tuning of large language models with higher training throughput. NeMo Evaluator provides simplified evaluation of AI models for custom benchmarks. NeMo Guardrails implements security controls to maintain compliance and appropriateness
 AI Paints A New Picture For The Future Of Art And DesignApr 26, 2025 am 11:10 AM
AI Paints A New Picture For The Future Of Art And DesignApr 26, 2025 am 11:10 AMAI: The Future of Art and Design Artificial intelligence (AI) is changing the field of art and design in unprecedented ways, and its impact is no longer limited to amateurs, but more profoundly affecting professionals. Artwork and design schemes generated by AI are rapidly replacing traditional material images and designers in many transactional design activities such as advertising, social media image generation and web design. However, professional artists and designers also find the practical value of AI. They use AI as an auxiliary tool to explore new aesthetic possibilities, blend different styles, and create novel visual effects. AI helps artists and designers automate repetitive tasks, propose different design elements and provide creative input. AI supports style transfer, which is to apply a style of image
 How Zoom Is Revolutionizing Work With Agentic AI: From Meetings To MilestonesApr 26, 2025 am 11:09 AM
How Zoom Is Revolutionizing Work With Agentic AI: From Meetings To MilestonesApr 26, 2025 am 11:09 AMZoom, initially known for its video conferencing platform, is leading a workplace revolution with its innovative use of agentic AI. A recent conversation with Zoom's CTO, XD Huang, revealed the company's ambitious vision. Defining Agentic AI Huang d
 The Existential Threat To UniversitiesApr 26, 2025 am 11:08 AM
The Existential Threat To UniversitiesApr 26, 2025 am 11:08 AMWill AI revolutionize education? This question is prompting serious reflection among educators and stakeholders. The integration of AI into education presents both opportunities and challenges. As Matthew Lynch of The Tech Edvocate notes, universit
 The Prototype: American Scientists Are Looking For Jobs AbroadApr 26, 2025 am 11:07 AM
The Prototype: American Scientists Are Looking For Jobs AbroadApr 26, 2025 am 11:07 AMThe development of scientific research and technology in the United States may face challenges, perhaps due to budget cuts. According to Nature, the number of American scientists applying for overseas jobs increased by 32% from January to March 2025 compared with the same period in 2024. A previous poll showed that 75% of the researchers surveyed were considering searching for jobs in Europe and Canada. Hundreds of NIH and NSF grants have been terminated in the past few months, with NIH’s new grants down by about $2.3 billion this year, a drop of nearly one-third. The leaked budget proposal shows that the Trump administration is considering sharply cutting budgets for scientific institutions, with a possible reduction of up to 50%. The turmoil in the field of basic research has also affected one of the major advantages of the United States: attracting overseas talents. 35
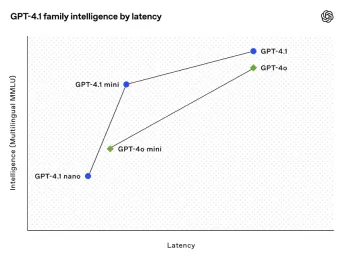 All About Open AI's Latest GPT 4.1 Family - Analytics VidhyaApr 26, 2025 am 10:19 AM
All About Open AI's Latest GPT 4.1 Family - Analytics VidhyaApr 26, 2025 am 10:19 AMOpenAI unveils the powerful GPT-4.1 series: a family of three advanced language models designed for real-world applications. This significant leap forward offers faster response times, enhanced comprehension, and drastically reduced costs compared t


Hot AI Tools

Undresser.AI Undress
AI-powered app for creating realistic nude photos

AI Clothes Remover
Online AI tool for removing clothes from photos.

Undress AI Tool
Undress images for free

Clothoff.io
AI clothes remover

Video Face Swap
Swap faces in any video effortlessly with our completely free AI face swap tool!

Hot Article
Hot Tools

mPDF
mPDF is a PHP library that can generate PDF files from UTF-8 encoded HTML. The original author, Ian Back, wrote mPDF to output PDF files "on the fly" from his website and handle different languages. It is slower than original scripts like HTML2FPDF and produces larger files when using Unicode fonts, but supports CSS styles etc. and has a lot of enhancements. Supports almost all languages, including RTL (Arabic and Hebrew) and CJK (Chinese, Japanese and Korean). Supports nested block-level elements (such as P, DIV),

SublimeText3 Chinese version
Chinese version, very easy to use

Notepad++7.3.1
Easy-to-use and free code editor

VSCode Windows 64-bit Download
A free and powerful IDE editor launched by Microsoft

SAP NetWeaver Server Adapter for Eclipse
Integrate Eclipse with SAP NetWeaver application server.

