
Home >Technology peripherals >AI >Published in the Nature sub-journal, the topological Transformer model predicts multi-scale protein-ligand interactions to assist drug development
Published in the Nature sub-journal, the topological Transformer model predicts multi-scale protein-ligand interactions to assist drug development

- 王林Original
- 2024-07-02 15:23:211221browse

A new artificial intelligence application will help researchers improve their drug development capabilities.
The project is called TopoFormer and was developed by an interdisciplinary team led by Professor Guowei Wei from the Department of Mathematics at Michigan State University.
TopoFormer transforms the three-dimensional information of a molecule into data that can be used by typical AI-based drug interaction models, extending the ability of these models to predict drug effectiveness.
“With artificial intelligence, you can make drug development faster, more efficient, and cheaper,” said Wei, who is also in the Department of Biochemistry and Molecular Biology and the Department of Electrical and Computer Engineering.
Professor Wei explained that in the United States, developing a drug takes about ten years and costs about $2 billion. Drug trials take up about half of the time, while the other half is spent discovering new treatment candidates to test.
TopoFormer has the potential to shorten development time. In this way, drug development costs can be reduced, thereby lowering drug prices for downstream consumers.
The study was titled "Multiscale topology-enabled structure-to-sequence transformer for protein–ligand interaction predictions" and was published in "Nature Machine Intelligence" on June 24, 2024.

"There are more than 20,000 proteins in our bodies." Wei said, "When a disease occurs, some or one of them becomes a target."
The first step, then, is to understand which or one of these proteins is affected by the disease. Which proteins. These proteins are also being targeted by researchers hoping to find molecules that can prevent, mitigate or counteract the effects of disease.
“When I have a goal, I try to find a large number of potential drugs for that specific goal,” Wei said.
Once scientists know which proteins a drug should target, they can feed the protein and the molecular sequence of the potential drug into a traditional computer model. These models can predict how drugs and targets will interact, guiding development and which drugs to test in clinical trials.
While these models can predict some interactions based solely on the chemical composition of the drug and protein, they also ignore important interactions that come from the shape and three-dimensional or 3D structure of the molecule.
Ibuprofen, discovered by chemists in the 1960s, is an example. There are two different ibuprofen molecules that have the same chemical sequence but slightly different 3D structures. Only one arrangement can bind to pain-related proteins and eliminate headaches.
Guowei Wei said: "Current deep learning models cannot explain the shape of drugs or proteins when predicting how they work together."
The Transformer architecture introduces a new technology that leverages the attention mechanism for cross-domain sequential data analysis. Inspired by this, Wei's team developed a topological Transformer model, TopoFormer, integrating Persistent Topological Hyperdigraph Laplacian (PTHL) with the Transformer framework.
Unlike traditional Transformers that handle protein and ligand sequences, TopoFormer inputs 3D protein-ligand complexes. It converts these complexes into sequences of topological invariants and homotopic shapes via PTHL, thereby capturing their physical, chemical, and biological interactions at multiple scales.
Pre-trained on diverse datasets, TopoFormer is able to understand complex molecular interactions, including stereochemical effects that are not apparent in the molecular sequence. Fine-tuning on a specific dataset can capture detailed interactions within a complex and their characteristics relative to the entire dataset, thereby enhancing downstream deep learning applications.
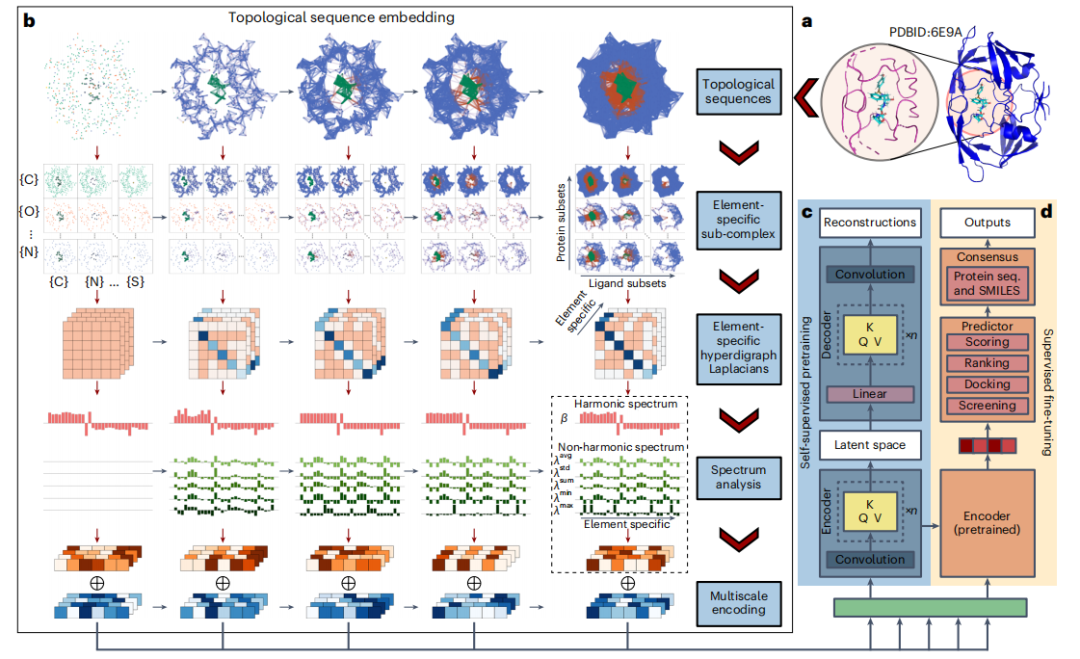
- To focus the analysis, researchers use a cutoff of 20 Å or, more precisely, 12 Å to identify reassortants and nearby protein atoms within a set distance.
- Then, TopoFormer converts the 3D molecular structure into a topological sequence through the topological sequence embedding module, using PTHL for multi-scale analysis. This process embeds various physical, chemical, and biological interactions into the vector sequence.
- TopoFormer uses unlabeled protein-ligand complexes for self-supervised pre-training and a Transformer encoder-decoder to reconstruct topological sequences. This stage prepares the model for understanding protein-ligand dynamics in the absence of labeled data by measuring accuracy by comparing output and input embeddings.
- After pre-training, TopoFormer enters the supervised fine-tuning stage of labeled complexes, where the initial embedding vectors become key features for downstream tasks such as scoring, ranking, docking, and screening. Each task has a dedicated header in the predictor module.
- To ensure accuracy and reduce bias, TopoFormer integrates multiple topological transformation deep learning models initialized with different seeds and complements them with sequence-based models.
- The final output is a consensus of these different predictions, making TopoFormer a comprehensive model for analyzing protein-ligand interactions, leveraging both topological insights and deep learning.
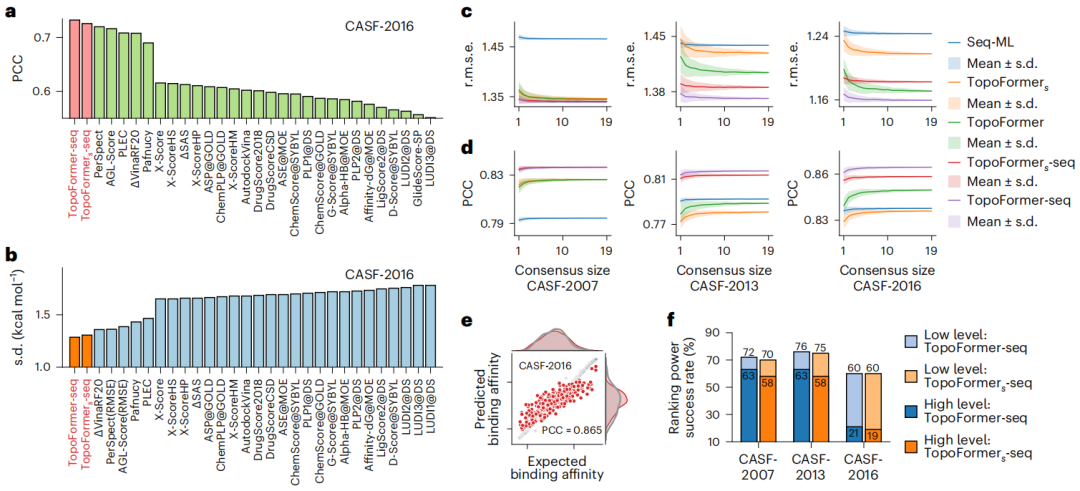
Illustration: TopoFormer’s performance in scoring and ranking tasks. (Source: paper)
This approach allows the use of directed hyperedges of various dimensions to model complex interactions beyond simple pairwise connections. Furthermore, the orientation of these edges combines physical and chemical properties, such as electronegativity and ionization energy, to provide a more nuanced representation than traditional methods. The researchers demonstrated this ability by distinguishing two B7C2H9 isomers with directed hyperedges, demonstrating the method's ability to effectively differentiate between elemental configurations.
When studying protein-ligand complexes, researchers adopt topological hyperdirected graphs as initial representations and further enhance them with PTHL theory to analyze their geometric and topological characteristics.
Drawing inspiration from physical systems such as molecular structures, where the zero-dimensional Hoch Laplacian operator is linked to the kinetic energy operator of the Hamiltonian of well-defined quantum systems, researchers extend the discrete analogy to topological super to the graph. These eigenvalues of the Laplacian matrix provide insight into the properties of topological objects, analogous to the energy spectrum of a physical system.
Compared to traditional persistent homology, the PTHL method marks a major advance by analyzing a wider range of structures beyond simplex complexes. It captures fundamental homology information and geometric insights, including Betti numbers and homotopic shape evolution, through the anharmonic spectrum of the persistent Laplacian operator.
The analysis results show that it provides a more comprehensive characterization compared to traditional homology. The multiplicity of zero eigenvalues of the Laplacian operator (corresponding to Betti's number) confirms that this method contains barcoding information, providing a powerful framework for understanding protein-ligand complexes.
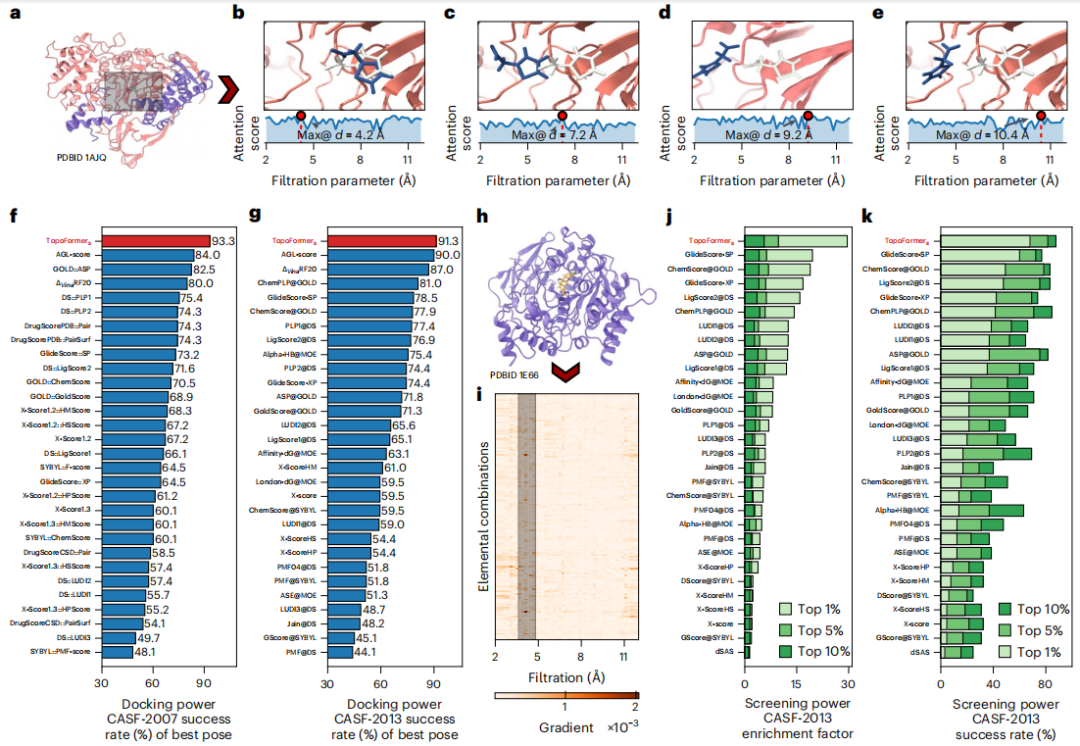
To capture the complex atomic interactions in protein-ligand complexes, including covalent, ionic, and van der Waals forces, the researchers used PTHL to perform multiscale analysis. This approach allows for the examination of cross-scale interactions by evolving topological sequences based on filtering parameters, thereby helping the Transformer model identify the weight of each scale on properties such as binding affinity.
Elemental interactions, including hydrogen bonding, van der Waals forces, and π stacking, are the basis for the stability and specificity of protein-ligand complexes. To analyze these interactions at the elemental level, the researchers introduced element-specific analysis in topological sequence embedding.
This method constructs sub-hypergraphs based on common heavy elements in proteins and ligands, generating element-specific Laplacian matrices to encode interactions within the complex. The technology extracts detailed physical and chemical features that enhance Transformer models' understanding of complex dynamics in protein-ligand interactions.
Conclusion
To summarize, TopoFormer is trained to read one form of information and convert it into another. In this case, it takes three-dimensional information about how proteins and drugs interact based on their shapes and reconstructs it into one-dimensional information that current models can understand.
The new model is trained on tens of thousands of protein-drug interactions, where each interaction between two molecules is recorded as a piece of code or a "word." These words are strung together to form a description of the drug-protein complex, creating a record of its shape.
"This way, you have many words strung together like sentences." Wei said.
Other models that predict new drug interactions can then read these sentences and provide them with more context. If a new drug were a book, TopoFormer can turn a rough story idea into a complete plot, ready to be written.
Paper link: https://www.nature.com/articles/s42256-024-00855-1
Related reports: https://phys.org/news/2024-06-drug-discovery-ai-3d -typical.html
The above is the detailed content of Published in the Nature sub-journal, the topological Transformer model predicts multi-scale protein-ligand interactions to assist drug development. For more information, please follow other related articles on the PHP Chinese website!