
Heim >Technologie-Peripheriegeräte >KI >Das Team der Shanghai Jiao Tong University entwickelt einen datengesteuerten Rahmen für aktives Lernen, um den Forschungsfortschritt bei Kohlenstoffnanomaterialien zu beschleunigen
Das Team der Shanghai Jiao Tong University entwickelt einen datengesteuerten Rahmen für aktives Lernen, um den Forschungsfortschritt bei Kohlenstoffnanomaterialien zu beschleunigen

- WBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBnach vorne
- 2024-01-16 22:33:191328Durchsuche
Herausgeber |
Obwohl die kontrollierte Synthese des katalytischen Substratwachstums zu Kohlenstoffnanostrukturen als vielversprechender Ansatz gilt, gibt es immer noch Herausforderungen bei dynamischen katalytischen Oberflächenwachstumsmechanismen und Designstrategien, die weiterer Forschung und Entwicklung bedürfen.
Kürzlich haben Forschungsteams der Shanghai Jiao Tong University und der Tohoku University in Japan die Wirksamkeit aktiver Modelle des maschinellen Lernens bei der Aufdeckung des mikroskopischen Prozesses des katalytischen Substratwachstums demonstriert. Durch die gemeinsame Anwendung von Molekulardynamik und Monte-Carlo-Methoden führten sie erfolgreich eine umfassende dynamische Simulation des Wachstums von Graphen auf Cu(111) durch. Um die Genauigkeit der Simulation zu verbessern, verwendete das Forschungsteam ein Gaußsches Näherungspotential. Diese Forschung liefert neue Werkzeuge und Methoden für ein tiefgreifendes Verständnis des katalytischen Wachstumsprozesses.
Durch diese Forschung haben wir eine praktische und effektive Methode abgeleitet, mit der Metall- oder Legierungssubstrate entworfen werden können, um die gewünschten Kohlenstoffnanostrukturen zu erhalten und weitere Reaktionsmöglichkeiten zu erkunden.
Die Forschung mit dem Titel „Aktives maschinelles Lernmodell für die dynamische Simulation und Wachstumsmechanismen von Kohlenstoff auf Metalloberflächen“ wurde am 6. Januar 2024 in „Nature Communications“ veröffentlicht.
... oder eine der vielversprechendsten Methoden für das kontrollierbare Wachstum kovalent gebundener Netzwerke aus dreidimensionalen Kohlenstoffatomen. Während Wachstumsmechanismen auf gewöhnlichen Oberflächen ausführlich untersucht wurden, ist das Wissen über die dynamischen und atomaren Faktoren, die die Graphenmasse auf Oberflächen mit hohem Index oder Verbundwerkstoffen steuern, begrenzt. Diese Forschungslücke hat die Entwicklung theoriegeleiteter Designansätze für neuartige katalytische Metallsubstrate beim Wachstum von Kohlenstoffnanostrukturen erheblich behindert.
Die experimentelle Suche nach Metall- oder Legierungskatalysatoren stellt aufgrund des breiten Spektrums potenzieller Substrate und der Empfindlichkeit des Kohlenstoffnanomaterial-Wachstumsprozesses gegenüber verschiedenen experimentellen Parametern erhebliche Herausforderungen dar. 
Um die Effizienz und Effektivität des dynamischen Trainings von Abscheidungsprozessen zu verbessern, ist ein genau definiertes Auswahlprotokoll erforderlich. Andererseits kann die Dynamik des Kohlenstoffwachstums auf metallischen Substraten durch wichtige seltene Ereignisse gesteuert werden. Daher bedarf es weiterer Forschung, wie die Trainingseffizienz von MLP durch die Kombination von Boosted-Sampling-Methoden mit klassischer Dynamik verbessert werden kann.
Datengesteuerter automatischer Lernrahmen zur Generierung von MLP mit minimalem Personalaufwand
Diese Studie schlägt einen datengesteuerten automatischen Lernrahmen zur Generierung von MLP mit minimalem Personalaufwand vor, der für das Kohlenstoffwachstum auf Metall- oder Legierungsoberflächen geeignet ist. Um diese Aufgabe zu lösen, verwendeten die Forscher (1) das Gaussian Approximation Potential (GAP)-Lernmodell (2) eine erweiterte Stichprobenmethode namens Time-Stamped Force Biased Monte Carlo (Time-Stamped Force Biased Monte Carlo). Monte Carlo, tfMC)-Methode zur Beschleunigung des Relaxationsprozesses nach der Kohlenstoffablagerung, wodurch wichtige seltene Ereignisse in die Trainingsdatenbank aufgenommen werden (3) Wählen Sie repräsentative Trainingsdaten basierend auf dem Smooth Overlap of Atomic Positions (SOAP)-Deskriptor aus (4); bewährte Carbon-Trainingssets; (5) automatisierte Screening-, Anpassungs- und Validierungsverfahren.
Abbildung 1: Schematische Darstellung des Kohlenstoffwachstums auf Metall-Machine-Learning-Potenzial (CGM-MLP), das durch dynamisches aktives Lernen während einer hybriden MD/tfMC-Simulation erzeugt wird. (Quelle: Papier)
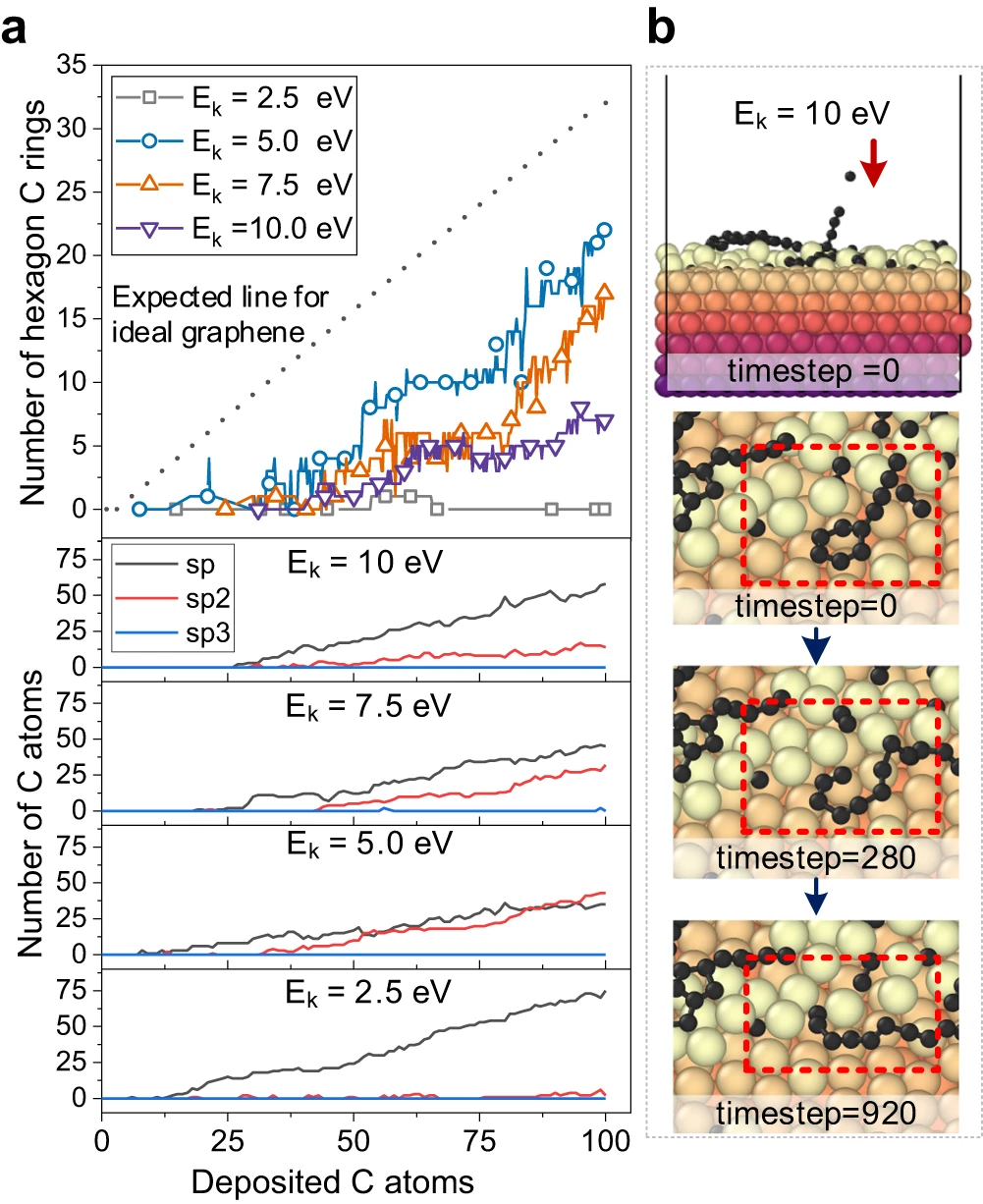
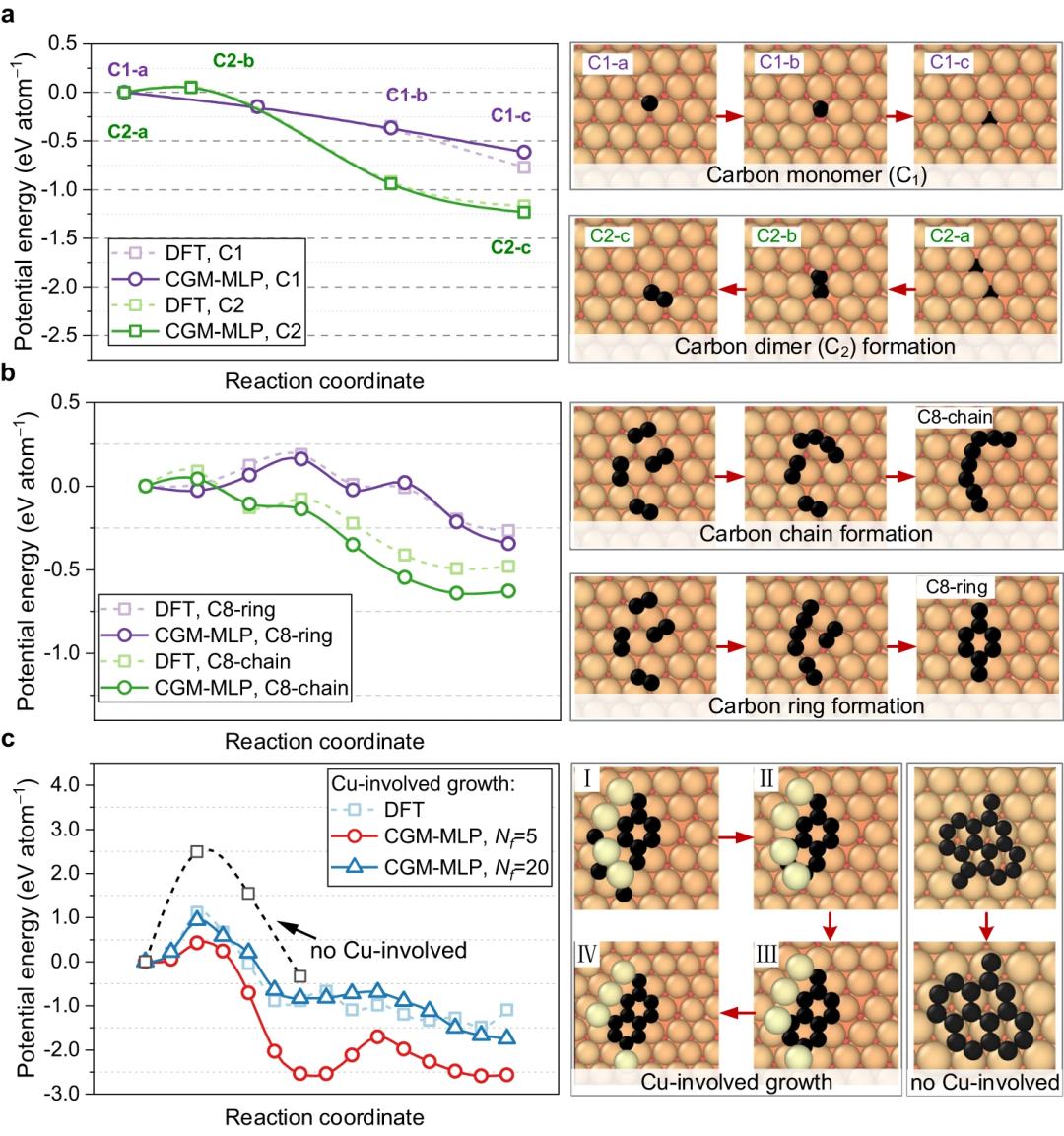
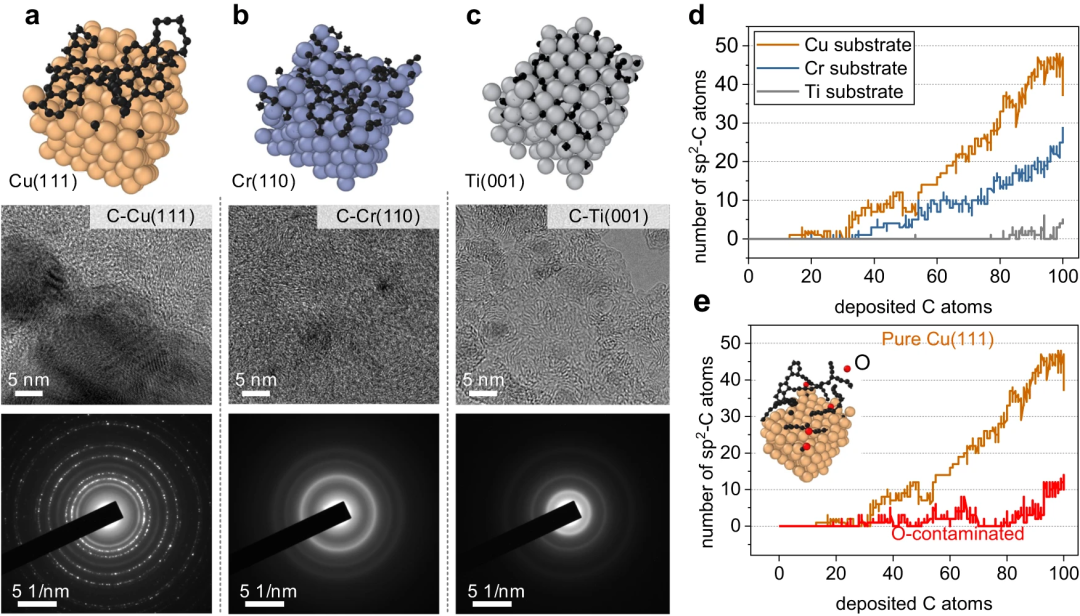
Erfolgreiche Nachbildung der Graphen-Keimbildung und des Kohlenstoffs auf Metalloberflächen durch Nutzung der hohen Genauigkeit des Carbon Growth Machine Learning Potential (CGM-MLP) und Einbeziehung seltener atomarer Ereignisse in die MD/tfMC-Methode. Die grundlegenden Teilprozesse im Zusammenhang mit Wachstum sind in der folgenden Abbildung dargestellt. Abbildung 2: CGM-MLP-gesteuerte Simulation des Graphenwachstums auf Cu(111) mit unterschiedlichen kinetischen Energien (Ek) des Kohlenstoffeinfalls. (Quelle: Papier) Das resultierende Potenzial wurde dann angewendet, um das Ablagerungswachstum von Kohlenstoffatomen auf der Cu (111)-Oberfläche zu untersuchen. Mit dieser Methode können die Schlüsselprozesse des Kohlenstoffwachstums auf Cu(111) korrekt erfasst werden, wie z. B. die Bildung und Migration von Kohlenstoffmonomeren und Oberflächendimeren unter der Oberfläche, die Entstehung eindimensionaler Kohlenstoffnanokristalle, die Graphenkeimbildung unter Beteiligung von Cu-Atomen und die Kantenpassivierung von Kohlenstoff Ketten und Niederschlagswachstumsprozess. Abbildung 3: Kohlenstoffstrukturanalyse und Beobachtung des Kohlenstoffringbruchs durch Hochenergiebeschuss. (Quelle: Papier) Abbildung 4: Minimale Energiepfade für Kohlenstoffdiffusion und Graphenkeimbildung, erhalten mit CGM-MLP auf Metall und DFT-basierten CI-NEB-Berechnungen (Crawling Image Nudged Elastic Band). (Quelle: Papier) Forscher simulierten die anfängliche Keimbildung auf verschiedenen Metalloberflächen, insbesondere die Kohlenstoffablagerung auf Cu(111), Cr(110), Ti(001) und O-kontaminiertem Cu(111), mit experimentellen Beobachtungen und DFT-Berechnungen Konstanz zeigen. Abbildung 5: Repräsentative Metalloberflächen für das Wachstum von Kohlenstoffnanostrukturen. (Quelle: Papier) Bedeutung der Forschung Zusammenfassend stellt diese Forschung einen bahnbrechenden Fortschritt bei der Integration von MLP und MD/tfMC dar und liefert übertragbare Informationen für die Gestaltung von Metall- oder Legierungssubstraten, um gewünschte Kohlenstoffnanostrukturen und eine effektive Strategie zu erhalten. CGM-MLP kombiniert effektiv die Genauigkeit von First-Principles-Methoden mit der Effizienz klassischer Kraftfelder. Die tfMC-Methode überwindet die zeitlichen Einschränkungen traditioneller AIMD- oder klassischer MD-Methoden. Darüber hinaus umfasst das automatisierte Trainings-Framework von CGM-MLP spezielle Abfragestrategien für den Aufbau dynamischer Trainingssätze in Ablagerungssimulationen und unterstreicht die Bedeutung der Berücksichtigung der lokalen Umgebung um abgelagerte Atome. Diese Fortschritte ermöglichen die direkte Untersuchung des Kohlenstoffwachstumsmechanismus auf komplexen Metalloberflächen. Das in dieser Studie vorgeschlagene, durch maschinelles Lernen gesteuerte Ablagerungsmodell bietet möglicherweise Möglichkeiten, das Wachstum einer Vielzahl von Kohlenstoffnanostrukturen (z. B. Graphen, Kohlenstoffnanoröhren, Graphit oder diamantähnliche Kohlenstofffilme) auf Metall- oder Legierungssubstraten mit mehreren Elementen zu untersuchen. 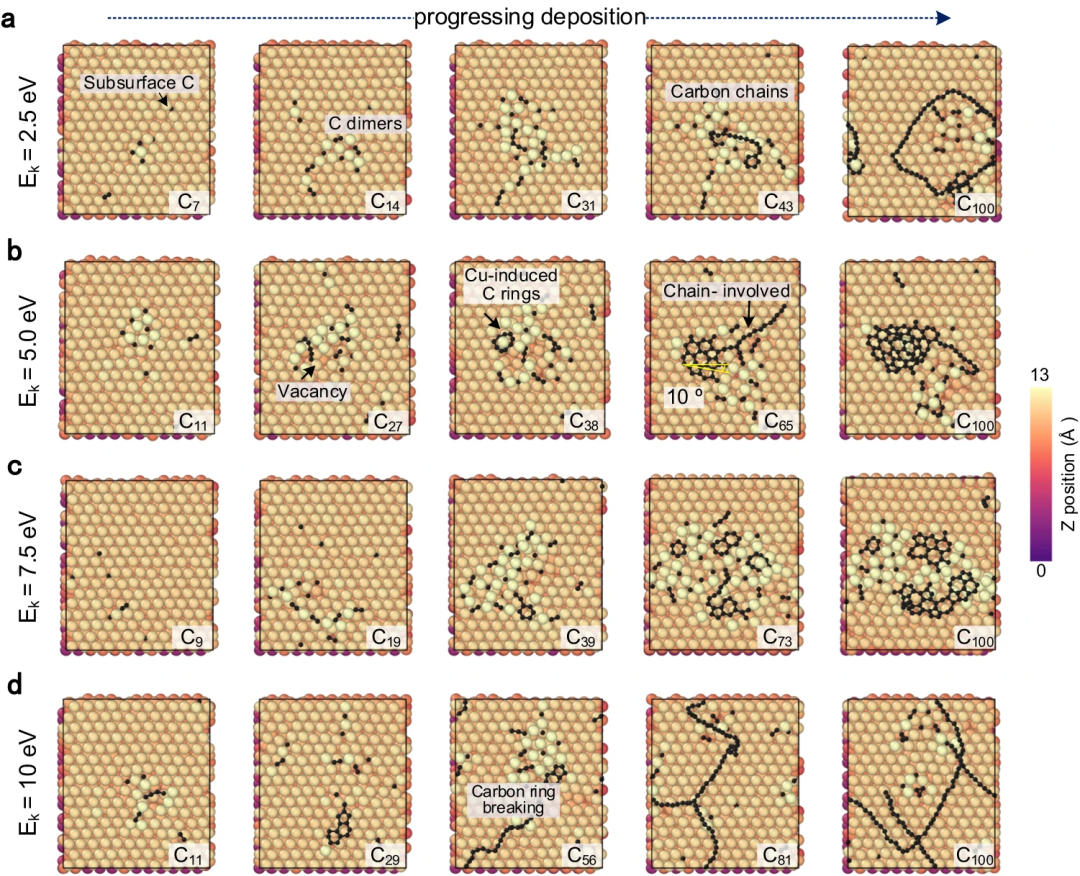
Das obige ist der detaillierte Inhalt vonDas Team der Shanghai Jiao Tong University entwickelt einen datengesteuerten Rahmen für aktives Lernen, um den Forschungsfortschritt bei Kohlenstoffnanomaterialien zu beschleunigen. Für weitere Informationen folgen Sie bitte anderen verwandten Artikeln auf der PHP chinesischen Website!