
Heim >Technologie-Peripheriegeräte >KI >KI wird nicht gelernt! Neue Forschungsergebnisse zeigen Wege auf, die Blackbox der künstlichen Intelligenz zu entschlüsseln
KI wird nicht gelernt! Neue Forschungsergebnisse zeigen Wege auf, die Blackbox der künstlichen Intelligenz zu entschlüsseln

- WBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBnach vorne
- 2024-01-16 21:30:31735Durchsuche
Künstliche Intelligenz (KI) hat sich rasant weiterentwickelt, doch für den Menschen sind leistungsstarke Modelle eine „Black Box“.
Wir verstehen das Innenleben des Modells und den Prozess, durch den es zu seinen Schlussfolgerungen gelangt, nicht.
Doch kürzlich gelang Professor Jürgen Bajorath, Cheminformatiker an der Universität Bonn, und seinem Team ein großer Durchbruch.
Sie haben eine Technik entwickelt, die zeigt, wie einige Systeme der künstlichen Intelligenz funktionieren, die in der Arzneimittelforschung eingesetzt werden.
Untersuchungen zeigen, dass Modelle der künstlichen Intelligenz die Wirksamkeit von Medikamenten hauptsächlich durch den Abruf vorhandener Daten vorhersagen, anstatt spezifische chemische Wechselwirkungen zu lernen.
—Mit anderen Worten, KI-Vorhersagen basieren ausschließlich auf dem Zusammensetzen von Erinnerungen, und maschinelles Lernen lernt nicht wirklich!
Ihre Forschungsergebnisse wurden kürzlich in der Zeitschrift Nature Machine Intelligence veröffentlicht.

Papieradresse: https://www.nature.com/articles/s42256-023-00756-9
In der Medizin suchen Forscher fieberhaft nach wirksamen Wirkstoffen zur Bekämpfung von Krankheiten —Welche Wirkstoffmoleküle sind am wirksamsten?
Üblicherweise werden diese wirksamen Moleküle (Verbindungen) an Proteine angedockt, die als Enzyme oder Rezeptoren fungieren und bestimmte physiologische Wirkungsketten auslösen.
In besonderen Fällen sind bestimmte Moleküle auch dafür verantwortlich, unerwünschte Reaktionen im Körper, wie zum Beispiel überschießende Entzündungsreaktionen, zu blockieren.
Die Anzahl der möglichen Verbindungen ist riesig und diejenige zu finden, die funktioniert, ist wie die Suche nach der Nadel im Heuhaufen.
Also nutzten die Forscher zunächst KI-Modelle, um vorherzusagen, welche Moleküle am besten andocken und sich stark an ihre jeweiligen Zielproteine binden würden. Diese Medikamentenkandidaten werden dann in experimentellen Studien noch detaillierter untersucht.
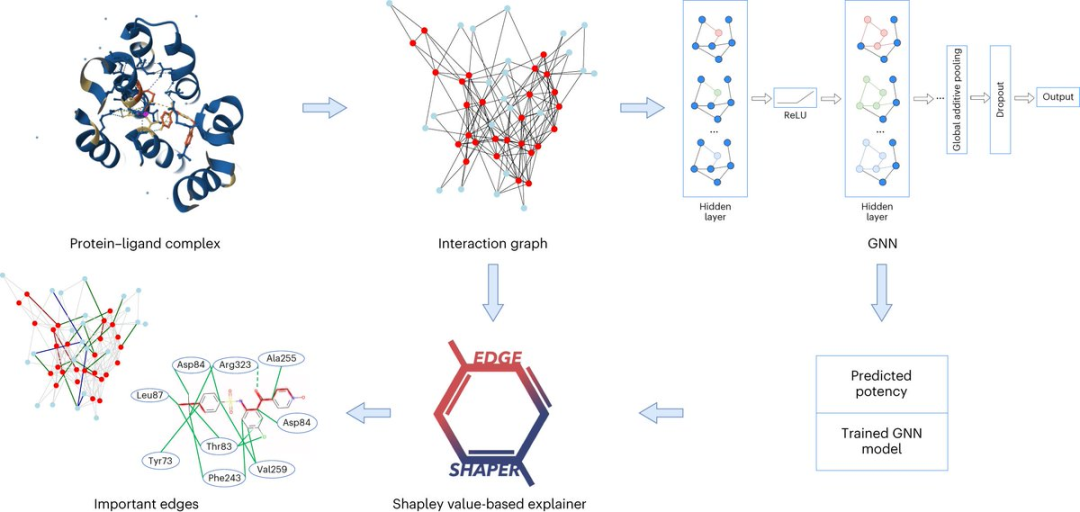
Seit der Entwicklung der künstlichen Intelligenz werden in der Arzneimittelforschung zunehmend KI-bezogene Technologien eingesetzt.
Graph Neural Network (GNN) eignet sich beispielsweise zur Vorhersage der Bindungsstärke eines bestimmten Moleküls an ein Zielprotein.
Ein Diagramm besteht aus Knoten, die Objekte darstellen, und Kanten, die Beziehungen zwischen Knoten darstellen. In der Diagrammdarstellung eines Protein-Ligand-Komplexes verbinden die Kanten des Diagramms Protein- oder Ligandenknoten und stellen die Struktur einer Substanz oder die Wechselwirkung zwischen einem Protein und einem Liganden dar.
Das GNN-Modell verwendet aus Röntgenstrukturen extrahierte Protein-Ligand-Interaktionskarten, um Ligandenaffinitäten vorherzusagen.
Professor Jürgen Bajorath sagte, dass das GNN-Modell für uns wie eine Blackbox ist und wir keine Möglichkeit haben zu wissen, wie es seine Vorhersagen ableitet.

Professor Jürgen Bajorath arbeitet am LIMES-Institut der Universität Bonn, am Bonn-Aachen International Center for Information Technology (Bonn-Aachen International Center for Information Technology) und am Lamarr Institute for Machine Learning and Artificial Intelligence (Lamarr-Institut für maschinelles Lernen und künstliche Intelligenz).
Wie funktioniert künstliche Intelligenz?
Forscher der Abteilung für Chemische Informatik der Universität Bonn haben gemeinsam mit Kollegen der Sapienza-Universität Rom detailliert analysiert, ob das graphische neuronale Netzwerk die Interaktion zwischen dem Protein und dem Liganden wirklich gelernt hat.
Die Forscher analysierten insgesamt sechs verschiedene GNN-Architekturen mit ihrer speziell entwickelten Methode „EdgeSHAPer“.
Das EdgeSHAPer-Programm kann feststellen, ob das GNN die wichtigsten Wechselwirkungen zwischen Verbindungen und Proteinen gelernt oder mit anderen Methoden Vorhersagen getroffen hat.
Die Wissenschaftler trainierten sechs GNNs mithilfe von Diagrammen, die aus den Strukturen von Protein-Ligand-Komplexen extrahiert wurden – wobei die Wirkungsweise der Verbindung und die Stärke ihrer Bindung an das Zielprotein bekannt waren.
Testen Sie dann das trainierte GNN an anderen Verbindungen und verwenden Sie EdgeSHAPer, um zu analysieren, wie das GNN Vorhersagen erzeugt.
„Wenn sich GNNs wie erwartet verhalten, müssen sie die Wechselwirkungen zwischen Verbindungen und Zielproteinen lernen und Vorhersagen treffen, indem sie bestimmte Wechselwirkungen priorisieren.“
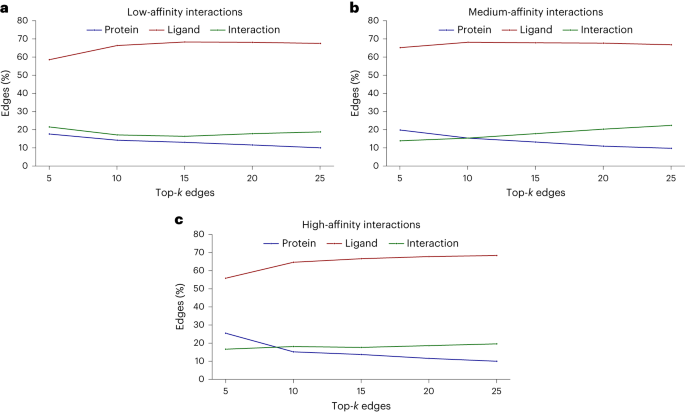
Der Analyse des Forschungsteams zufolge gelang dies jedoch sechs GNNs grundsätzlich nicht. Die meisten GNNs lernen nur einige Protein-Arzneimittel-Wechselwirkungen und konzentrieren sich hauptsächlich auf Liganden.
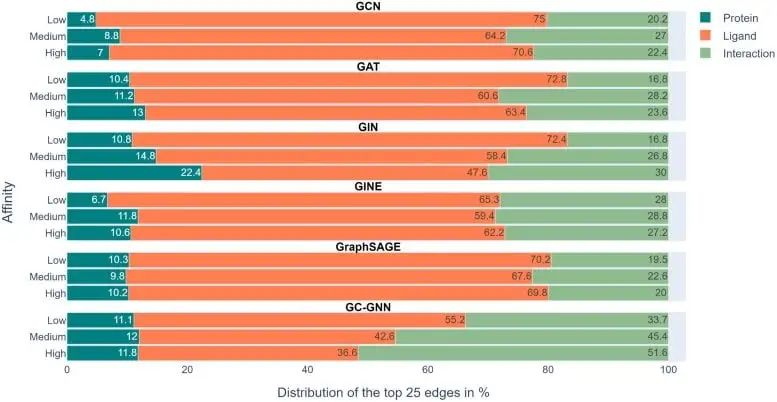
Die obige Abbildung zeigt die experimentellen Ergebnisse in 6 GNNs. Der Farbskalenbalken stellt den durchschnittlichen Anteil von Proteinen, Liganden und Wechselwirkungen in den oberen 25 Kanten jeder von EdgeSHAPer ermittelten Vorhersage dar.
Wir können sehen, dass das Modell die durch Grün dargestellte Interaktion lernen muss, ihr Anteil im gesamten Experiment jedoch nicht hoch ist und der orangefarbene Balken, der den Liganden darstellt, den größten Anteil ausmacht.
Um die Bindungsstärke eines Moleküls an ein Zielprotein vorherzusagen, „merken“ sich Modelle in erster Linie die chemisch ähnlichen Moleküle und ihre Bindungsdaten, auf die sie während des Trainings gestoßen sind, unabhängig vom Zielprotein. Diese erinnerten chemischen Ähnlichkeiten bestimmen im Wesentlichen die Vorhersage.
Das erinnert an den „Clever-Hans-Effekt“ – genau wie das Pferd, das scheinbar zählen kann, sich aber tatsächlich auf die Nuancen der Mimik und Gestik seiner Gefährten stützt, um auf das Erwartete zu schließen Ergebnisse.
Dies kann bedeuten, dass die sogenannte „Lernfähigkeit“ von GNN möglicherweise unhaltbar ist und die Vorhersagen des Modells weitgehend überschätzt werden, da chemische Kenntnisse und einfachere Methoden verwendet werden können, um Vorhersagen mit derselben Qualität durchzuführen.
Allerdings wurde in der Studie auch ein weiteres Phänomen entdeckt: Wenn die Wirksamkeit der Testverbindung zunimmt, neigt das Modell dazu, mehr Interaktionen zu lernen.
Vielleicht können diese GNNs durch Modifizierung der Darstellungs- und Trainingstechniken weiter in die gewünschte Richtung verbessert werden. Allerdings ist die Annahme, dass physikalische Größen aus molekularen Graphen gelernt werden können, grundsätzlich mit Vorsicht zu genießen.
„Künstliche Intelligenz ist keine schwarze Magie.“
Das obige ist der detaillierte Inhalt vonKI wird nicht gelernt! Neue Forschungsergebnisse zeigen Wege auf, die Blackbox der künstlichen Intelligenz zu entschlüsseln. Für weitere Informationen folgen Sie bitte anderen verwandten Artikeln auf der PHP chinesischen Website!