
Heim >Technologie-Peripheriegeräte >KI >KI-Alchemie revolutioniert die Chemie: MIT-Wissenschaftler nutzen generative KI, um in sechs Sekunden neue chemische Reaktionen zu erzeugen
KI-Alchemie revolutioniert die Chemie: MIT-Wissenschaftler nutzen generative KI, um in sechs Sekunden neue chemische Reaktionen zu erzeugen

- WBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBnach vorne
- 2023-12-18 12:49:10542Durchsuche

Was neu geschrieben werden muss, ist: Herausgeber | Kaixia
Chemie, ausgehend von der alten Alchemie des „äquivalenten Austauschs“, war schon immer eine Disziplin, die die Wechselwirkung zwischen Substanzen untersucht und kontrolliert. Durch die kontinuierliche Erschließung und Nutzung neuer chemischer Reaktionen wurden viele neue Materialien entwickelt. Diese neuen Materialien machen das Leben der Menschen nicht nur komfortabler, sondern verbessern auch die Effizienz der Energienutzung und fördern eine nachhaltige Entwicklung. Eine grundlegende chemische Reaktion besteht aus Reaktanten, Übergangszuständen (TS) und Produkten. Übergangszustände sind entscheidende 3D-Strukturen in der Chemie und werden häufig verwendet, um chemische Reaktionsmechanismen zu verstehen, Reaktionsenergiebarrieren abzuschätzen und riesige Reaktionsnetzwerke zu erkunden. Aufgrund der extrem kurzen Zeit (Femtosekunden-Ordnung), in der sie während der Reaktion vorliegen, ist es jedoch nahezu unmöglich, den Übergangszustand experimentell zu isolieren und zu charakterisieren.
Umgeschriebener Inhalt: Normalerweise verwenden Menschen quantenchemische Berechnungsmethoden, um den Übergangszustand zwischen bekannten Reaktanten und Produkten durch wiederholtes Lösen der Schrödinger-Gleichung zu bestimmen. Allerdings ist diese Berechnungsmethode sehr teuer und bekannt für ihre häufigen Fehler. Gleichzeitig ist diese Methode durch persönliche Erfahrung, Intuition und Rechenressourcen begrenzt, und auch die chemischen Reaktionen, die jeder Mensch erforschen kann, sind begrenzt. Diese Einschränkung ist besonders fatal, wenn unbekannte komplexe Reaktionen untersucht werden. Dies führt dazu, dass Forscher einige potenzielle Reaktionen ignorieren und dadurch den Reaktionsmechanismus falsch einschätzen, was sich auf das Design katalytischer Materialien auswirkt
Als Reaktion auf dieses Problem entwickelte eine Forschergruppe des Massachusetts Institute of Technology (MIT) eine darauf basierende alternative Methode Maschinelles Lernen kann diese Strukturen in Sekundenschnelle entdecken. Ihr neues Modell könnte Chemikern dabei helfen, neue Reaktionen und Katalysatoren zu erforschen und zu entwerfen, um nützliche Produkte mit hohem Mehrwert zu erzeugen, etwa Kraftstoffverbindungen oder Arzneimittel. Darüber hinaus ist das Modell in der Lage, natürlich vorkommende chemische Reaktionen zu simulieren, die beispielsweise für die Entwicklung des Lebens auf der frühen Erde von entscheidender Bedeutung sind.
Heather Kulik, Professorin für Chemieingenieurwesen und Chemie am MIT, wies darauf hin, dass das Verständnis der spezifischen Struktur des Übergangszustands sehr wichtig ist, um Katalysatoren zu entwerfen oder zu verstehen, wie natürliche Systeme bestimmte Transformationen durchführen.
Verwandte Forschungsarbeiten tragen den Titel „Genaue Übergangszustände“. Generation mit einem objektbewussten „Equivariant Elementary Reaction Diffusion Model“ wurde in der internationalen Top-Zeitschrift „Nature Computational Science“ veröffentlicht.
Dr. Duan Chenru vom MIT ist der erste Autor des Artikels, ein Doktorand an der Cornell University, und Professor Heather Kulik vom MIT . Ursprünglicher Link: [https://rdcu.be/dtGSF]
 Bitte klicken Sie auf den folgenden Link, um das Papier anzuzeigen: https://www.nature.com/articles/s43588-023-00563-7
Bitte klicken Sie auf den folgenden Link, um das Papier anzuzeigen: https://www.nature.com/articles/s43588-023-00563-7
MIT College News berichtete auch über diese Studie.
 Link zur Berichterstellung: https://news.mit.edu/2023/computational-model-captures-elusive-transition-states-1215 ist: Theoretische Schwierigkeiten
Link zur Berichterstellung: https://news.mit.edu/2023/computational-model-captures-elusive-transition-states-1215 ist: Theoretische Schwierigkeiten
Um das Problem der langen Berechnungszeit zu lösen, haben einige Forscher kürzlich damit begonnen, maschinelles Lernen einzusetzen Modelle zur Entdeckung von Übergangszustandsstrukturen. Allerdings erfordern fast alle bisher entwickelten Modelle, dass die beiden Reaktanten als Ganzes modelliert werden, wobei die Reaktanten relativ zueinander eine bestimmte geometrische Konfiguration beibehalten. Jede andere mögliche Konfiguration wird vom Modell des maschinellen Lernens als neue Reaktion verwechselt. Dr. Duan Chenru sagte, dass die Reaktantenmoleküle im Prinzip immer noch die gleiche chemische Reaktion vor und nach der Rotation erfahren können. Genau wie wenn wir über die Elektrolyse von Wasser sprechen, sagen wir nur, dass Wasser unter bestimmten Bedingungen in Sauerstoff und Wasserstoff umgewandelt wird, ohne die relativen geometrischen Positionen dieser Moleküle zu beschreiben. Bei herkömmlichen Methoden des maschinellen Lernens behandelt das Modell die Reaktionen von Reaktanten und Produkten an unterschiedlichen geometrischen Positionen jedoch als zwei unterschiedliche Reaktionen. Dadurch wird das Training des maschinellen Lernens schwieriger und die Genauigkeit nimmt ab
Das Diffusionsmodell ist ein generatives Modell, das in der Bildverarbeitung weit verbreitet ist. In jüngster Zeit werden Diffusionsmodelle auch zur Generierung von 3D-Molekül- und Proteinstrukturen, zum Protein-Ligand-Docking und zum strukturbasierten Arzneimitteldesign eingesetzt. In diesen Anwendungen verwenden Diffusionsmodelle 3D-Graph-Neuronale Netze (GNNs) der speziellen euklidischen Gruppe (SE(3)), um die Ausrichtung, Translations- und Rotationssymmetrien von Molekülen zu bewahren. Elementarreaktionen bestehen jedoch aus Reaktanten, Übergangszuständen und Produkten und folgen der „objektbewussten“ SE(3)-Symmetrie. Dies liegt daran, dass die Wechselwirkung zwischen den drei Objekten in der Elementarreaktion nicht im 3D-Euklidischen Raum stattfindet, sondern ein kausaler Zusammenhang auf der höherdimensionalen elektronischen potentiellen Energieoberfläche ist. Daher kann das bestehende Diffusionsmodell, das auf SE(3) GNN basiert, aufgrund der Zerstörung der Symmetrie Probleme haben . (Quelle: Paper)
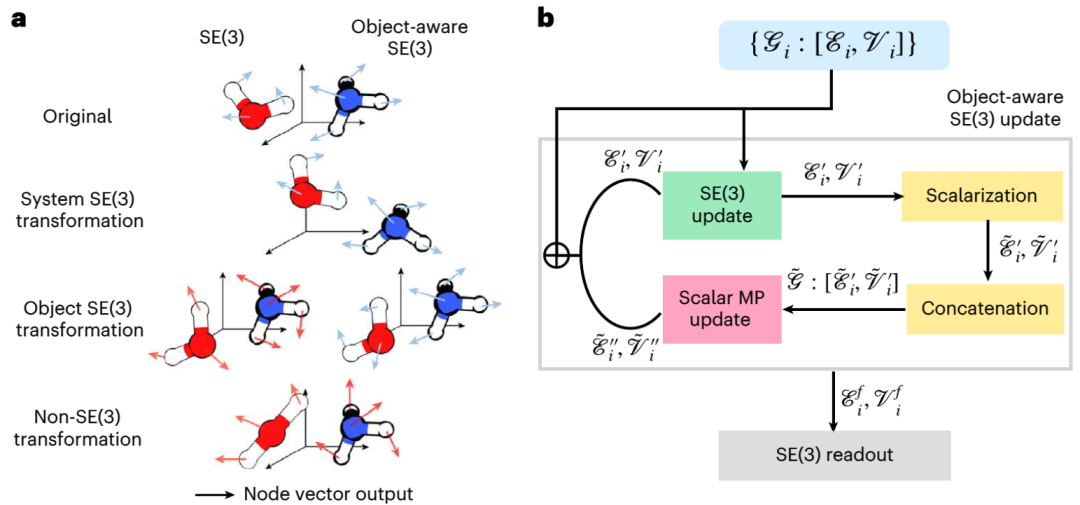 Lösung
Lösung
Abbildung: Übersicht über das äquivariante Diffusionsmodell (EDM) zur Generierung von Proben molekularer Systeme. (Quelle: Papier)
In der Studie verwendeten Forscher Quantencomputermethoden, um die Strukturen von Reaktanten, Übergangszuständen und Produkten von 9.000 verschiedenen chemischen Reaktionen im Trainingssatz zu ermitteln. Außerdem wurden etwa 1.000 bisher unbekannte Reaktionen getestet, was die Generierung von 40 möglichen Strukturen für jeden Übergangszustand erforderte. Auf dieser Grundlage führten die Forscher in Kombination mit Unsicherheitsschätzungen nur quantenchemische Berechnungen für die 14 % der Reaktionen mit der höchsten Modellunsicherheit durch und erreichten dabei erfolgreich einen durchschnittlichen absoluten Fehler von 2,6 kcal/mol. Dies ermöglicht Ergebnisse innerhalb einer Größenordnung Fehler bei der Verwendung von OA-ReactDiff zur Schätzung der Reaktionsraten bei 300 °C. Verglichen mit der durch quantenchemische Berechnungen ermittelten Übergangszustandsstruktur liegt der mittlere quadratische Fehler (RMSD) der durch OA-ReactDiff erzeugten Struktur im Bereich von 0,06 Angström (sechs Tausendstel Nanometer), eine Fehlergröße, die nahezu nicht zu unterscheiden ist mit bloßem Auge sichtbar
Was noch erfreulicher ist, ist, dass OA-ReactDiff nur 6 Sekunden benötigt, um eine Übergangszustandsstruktur zu erzeugen, was mindestens 1000-mal schneller ist als quantenchemische Berechnungen. Dadurch erreicht der Algorithmus erfolgreich eine extrem hohe Genauigkeit und Schnelligkeit bei der Berechnung von TS-Strukturen und Reaktionsenergiebarrieren. 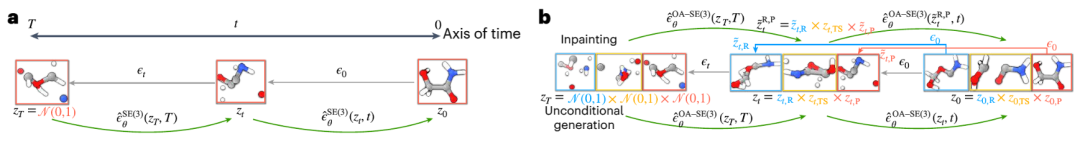
Abbildung: Bewertung der strukturellen Ähnlichkeit zwischen der von OA-ReactDiff generierten TS-Struktur und der realen TS-Struktur. (Quelle: Papier)
Professor Kulik beklagte auch: „Wir konnten uns kaum vorstellen, dass mit nur einem Gedanken Tausende von Übergangszuständen erzeugt werden könnten
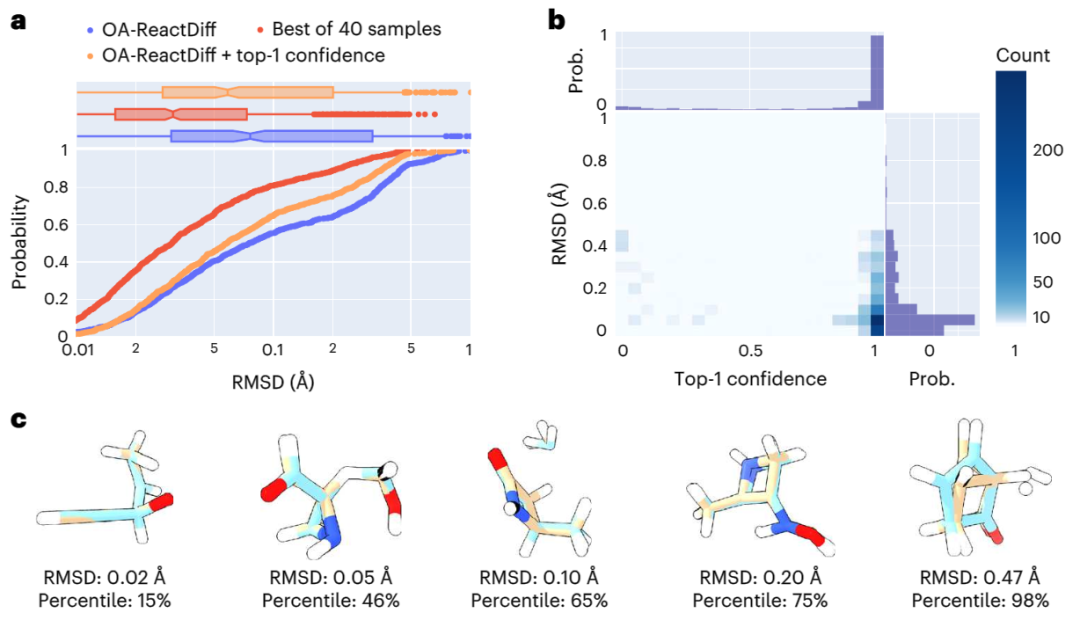 Der Inhalt, der neu geschrieben werden muss, ist: Illustration: OA-ReactDiff plus empfiehlt die Energieleistung von TS-Strukturen. (Quelle: Paper)
Der Inhalt, der neu geschrieben werden muss, ist: Illustration: OA-ReactDiff plus empfiehlt die Energieleistung von TS-Strukturen. (Quelle: Paper)
Was neu geschrieben werden muss, ist: Diese Forschung ist die erste, die ein 3D-Diffusionsmodell bei chemischen Reaktionen verwendet. Die Bedeutung dieser Arbeit kann nicht ignoriert werden, obwohl die Forscher nur Verbindungen mit einer geringeren Anzahl von Atomen (
Professor Kulik wies darauf hin: „Selbst wenn es um größere Systeme oder sogar enzymkatalysierte Systeme geht, ist es immer noch möglich, Informationen über die verschiedenen Arten zu erhalten, auf denen sich Atome am wahrscheinlichsten neu anordnenDie Forscher planen nun, ihr Modell um weitere Komponenten, beispielsweise Katalysatoren, zu erweitern. OA-ReactDiff nutzt die Zufälligkeit generativer KI und kann unerwartete chemische Reaktionen untersuchen. Diese Funktion ergänzt das bestehende, auf Chemie basierende, intuitive Reaktionsexplorations-Framework, hilft beim Aufbau eines vollständigeren chemischen Reaktionsnetzwerks und hilft bei der Entwicklung und dem Design neuer katalytischer Materialien. Die Forschung in diesem Bereich kann ihnen dabei helfen, die Entdeckung neuer Katalysatoren für bestimmte Reaktionen zu beschleunigen. Darüber hinaus könnte der von ihnen vorgeschlagene Algorithmus für die Entwicklung neuer Prozesse für Arzneimittel, Kraftstoffe oder andere nützliche Verbindungen nützlich sein, insbesondere wenn die Synthese viele chemische Schritte umfasst.
Dr. Duan Chenru wies darauf hin, dass alle diese Berechnungen in der Vergangenheit mit quantenchemischen Methoden durchgeführt wurden, aber jetzt können wir die Quantenchemie durch schnellere generative Modelle ersetzen
Die Forscher wiesen auch darauf hin, dass chemische Reaktionen den Kern der chemischen Forschung bilden . Neben dem Katalysatordesign, das auf industrielle Anwendungen ausgerichtet ist, bietet OA-ReactDiff auch viele interessante potenzielle Anwendungen, wie die Erforschung von Gaswechselwirkungen, die auf anderen Planeten auftreten können, die Simulation von Reaktionsprozessen während der Entwicklung des frühen Lebens auf der Erde usw.
Das obige ist der detaillierte Inhalt vonKI-Alchemie revolutioniert die Chemie: MIT-Wissenschaftler nutzen generative KI, um in sechs Sekunden neue chemische Reaktionen zu erzeugen. Für weitere Informationen folgen Sie bitte anderen verwandten Artikeln auf der PHP chinesischen Website!