
Heim >Technologie-Peripheriegeräte >KI >Weltneuheit: Der neue KI-Algorithmus von Molecular Heart zur Überwindung der Probleme der Proteinseitenkettenvorhersage und des Sequenzdesigns
Weltneuheit: Der neue KI-Algorithmus von Molecular Heart zur Überwindung der Probleme der Proteinseitenkettenvorhersage und des Sequenzdesigns

- 王林nach vorne
- 2023-06-06 13:20:231228Durchsuche
Maschinenherzsäule
Machine Heart Redaktion
PSCP Deep Architecture AttnPacker – stark optimierter KI-Algorithmus.
Die Bildung der Proteinstruktur und -funktion hängt weitgehend von der Wechselwirkung zwischen Seitenkettenatomen ab. Daher ist die genaue Proteinseitenkettenvorhersage (PSCP) ein Schlüsselelement bei der Lösung der Probleme der Proteinstrukturvorhersage und des Proteindesigns. Frühere Proteinstrukturvorhersagen konzentrierten sich jedoch hauptsächlich auf die Hauptkettenstruktur, und die Vorhersage der Seitenkettenstruktur war schon immer ein schwieriges Problem, das nicht vollständig gelöst wurde.
Kürzlich hat das Team von Xu Jinbo bei Molecular Heart einen neuen AttnPacker mit PSCP-Deep-Architektur auf den Markt gebracht, der erhebliche Verbesserungen in Bezug auf Geschwindigkeit, Speichereffizienz und Gesamtgenauigkeit erzielt hat. Es ist derzeit der bekannteste Algorithmus zur Vorhersage der Seitenkettenstruktur und der erste in Ein KI-Algorithmus, der gleichzeitig eine Proteinseitenkettenvorhersage und ein Sequenzdesign durchführen kann.
Das Papier wurde in den Proceedings of the National Academy of Sciences (PNAS) veröffentlicht und sein Pre-Training-Modell, Quellcode und Inferenzskripte wurden als Open Source auf Github bereitgestellt.

Papierlink:
https://www.pnas.org/doi/10.1073/pnas.2216438120#supplementary-materials
Open-Source-Link:
https://github.com/MattMcPartlon/AttnPacker
Hintergrund
Protein wird aus mehreren Aminosäuren gefaltet und ist in seiner Struktur in Hauptkette und Seitenkette unterteilt. Unterschiede in den Seitenketten haben einen großen Einfluss auf die Struktur und Funktion von Proteinen, insbesondere auf die biologische Aktivität. Basierend auf einem klaren Verständnis der Seitenkettenstruktur können Wissenschaftler die dreidimensionale Struktur von Proteinen genauer bestimmen, Protein-Protein-Wechselwirkungen analysieren und rationales Proteindesign durchführen. Bei der Anwendung auf dem Gebiet des Arzneimitteldesigns können Wissenschaftler schnell und genauer geeignete Bindungsstellen für Arzneimittel und Rezeptoren finden und bei Bedarf sogar Bindungsstellen optimieren oder entwerfen. Im Bereich der Enzymoptimierung können Wissenschaftler Sequenzen optimieren, die mehrere Seitenketten ermöglichen an katalytischen Reaktionen teilzunehmen, um effizientere und spezifischere katalytische Effekte zu erzielen.
Die meisten aktuellen Algorithmen zur Vorhersage der Proteinstruktur konzentrieren sich hauptsächlich auf die Strukturanalyse der Hauptkette, aber die Vorhersage der Proteinseitenkettenstruktur ist immer noch ein Problem, das nicht vollständig gelöst wurde. Unabhängig davon, ob es sich um beliebte Algorithmen zur Vorhersage der Proteinstruktur wie AlphaFold2 oder um Algorithmen mit Fokus auf die Vorhersage der Seitenkettenstruktur wie DLPacker und RosettaPacker handelt, sind weder die Genauigkeit noch die Geschwindigkeit zufriedenstellend. Dies bringt auch Einschränkungen für das Proteindesign mit sich.
Traditionelle Methoden wie RosettaPacker verwenden hauptsächlich Methoden zur Energieoptimierung, bei denen zunächst die Verteilung der Seitenkettenatome gruppiert wird und dann die Seitenkettengruppierungen nach einer bestimmten Aminosäure durchsucht werden, um die Kombination mit der kleinsten Energie zu finden. Diese Methoden unterscheiden sich in erster Linie von der Auswahl der Rotamer-Bibliotheken, Energiefunktionen und Energieminimierungsverfahren durch den Forscher, wobei die Genauigkeit durch die Verwendung von Suchheuristiken und diskreten Stichprobenverfahren begrenzt ist. In der Branche gibt es auch Side-Chain-Vorhersagemethoden, die auf Deep Learning basieren, beispielsweise DLPacker, das PSCP als Bild-zu-Bild-Konvertierungsproblem formuliert und eine U-Net-Modellstruktur übernimmt. Allerdings sind die Vorhersagegenauigkeit und -geschwindigkeit noch nicht optimal.
Methode
AttnPacker ist eine End-to-End-Deep-Learning-Methode zur Vorhersage von Proteinseitenkettenkoordinaten. Es simuliert gemeinsam Seitenkettenwechselwirkungen mit direkt vorhergesagten Seitenkettenstrukturen, die physikalisch besser realisierbar sind, mit weniger Atomkollisionen und idealeren Bindungslängen und -winkeln.
Insbesondere führt AttnPacker eine Tiefenkartenkonverterarchitektur ein, die die geometrischen und relationalen Aspekte von PSCP nutzt. Inspiriert von AlphaFold2 schlägt Molecular Heart positionsbewusste Dreiecksaktualisierungen vor, um paarweise Merkmale mithilfe eines graphbasierten Frameworks zur Berechnung der Dreiecksaufmerksamkeit und multiplikativer Aktualisierungen zu optimieren. Mit diesem Ansatz verfügt AttnPacker über deutlich weniger Speicher und ein Modell mit höherer Kapazität. Darüber hinaus untersucht Molecular Heart mehrere SE (3) äquivariante Aufmerksamkeitsmechanismen und schlägt eine äquivariante Transformatorarchitektur für das Lernen aus 3D-Punkten vor.
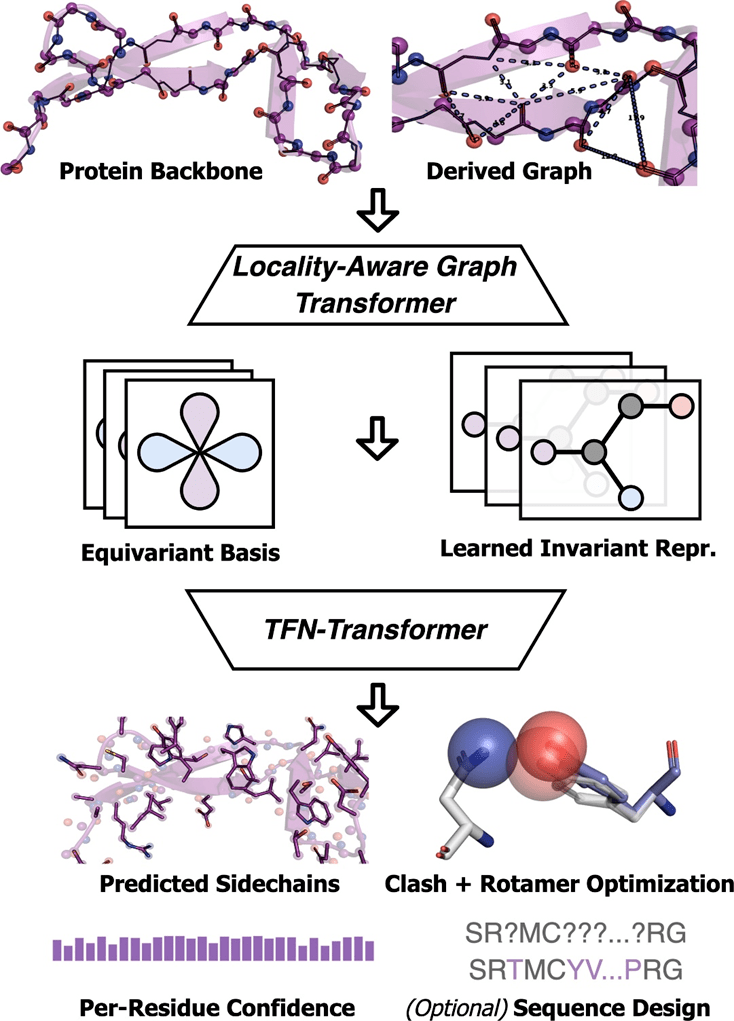
AttnPacker führt den Prozess aus. Die Koordinaten und die Sequenz des Proteinrückgrats werden als Eingabe verwendet, und die räumliche Merkmalskarte und die Äquivariablenbasis werden basierend auf den Koordinateninformationen abgeleitet. Die Feature-Map wird vom invarianten Graph-Transformer-Modul verarbeitet und dann an einen äquivarianten TFN-Transformer übergeben, der vorhergesagte Seitenkettenkoordinaten, Konfidenzwerte für jeden Rest und optionale Designsequenzen ausgibt. Die vorhergesagten Koordinaten werden nachbearbeitet, um alle räumlichen Konflikte zu beseitigen und eine idealisierte Geometrie sicherzustellen.
Effekt
In Bezug auf die Vorhersageleistung zeigt AttnPacker Verbesserungen in der Genauigkeit und Effizienz sowohl für natürliche als auch für nicht-natürliche Backbone-Strukturen. Gleichzeitig ist die physikalische Machbarkeit gewährleistet, Abweichungen von idealen Bindungslängen und -winkeln sind vernachlässigbar und es entsteht nur eine minimale atomare sterische Hinderung.
Molecular Heart führt Vergleichstests mit AttnPacker und den aktuellen hochmodernen Methoden – SCWRL4, FASPR, RosettaPacker und DLPacker – an den natürlichen und nicht-nativen Protein-Backbone-Datensätzen CASP13 und CASP14 durch. Die Ergebnisse zeigen, dass AttnPacker herkömmliche Methoden zur Vorhersage von Proteinseitenketten auf CASP13- und CASP14-nativen Backbones deutlich übertrifft, wobei die durchschnittlichen Rekonstruktions-RMSDs bei jedem Testsatz um mehr als 18 % niedriger sind als bei der suboptimalen Methode. AttnPacker übertrifft auch die Deep-Learning-Methode DLPacker, indem es den durchschnittlichen RMSD um mehr als 11 % reduziert und gleichzeitig die Genauigkeit der Sidechain-Dieder deutlich verbessert. Zusätzlich zur Genauigkeit weist AttnPacker deutlich weniger Atomkollisionen auf als andere Methoden.
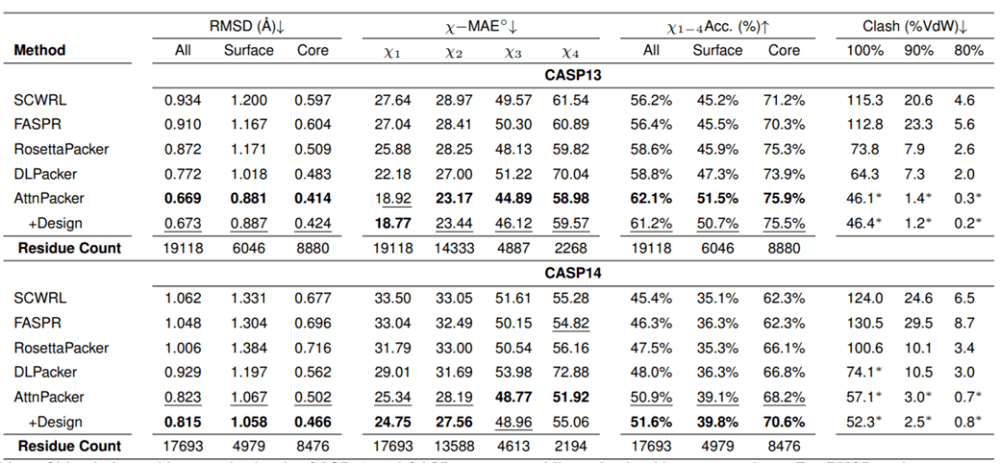
Angesichts der natürlichen Rückgratstruktur werden die Ergebnisse der Vorhersage der Seitenkettenstruktur jedes Algorithmus für die Zielproteine CASP13 und CASP14 angegeben. Sternchen zeigen an, dass die durchschnittlichen Konfliktwerte niedriger sind als die native Struktur – 56,0, 5,9 und 0,4 für CASP13 und 80,4, 7,9 und 2,5 für CASP14.
Auf nicht-nativen CASP13- und CASP14-Backbones ist AttnPacker auch deutlich besser als andere Methoden, und auch atomare Kollisionen sind deutlich geringer als bei anderen Methoden.
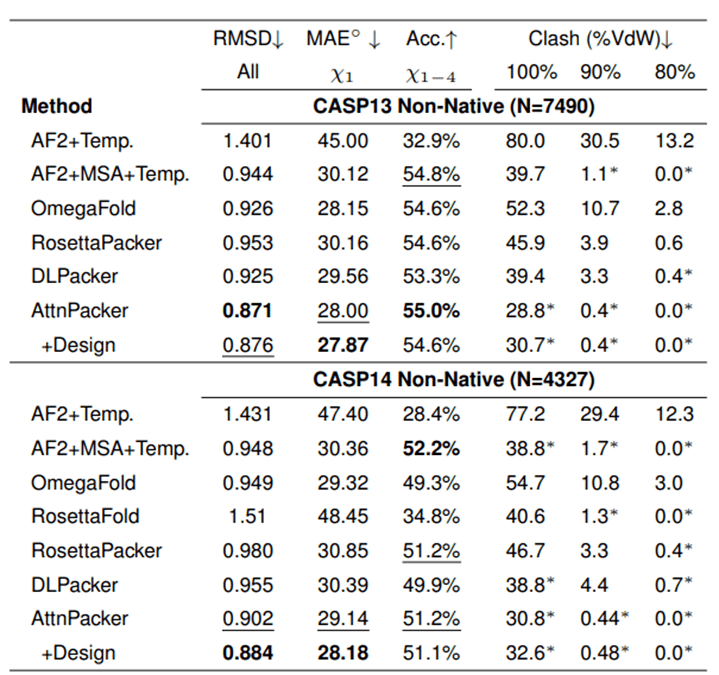
Angesichts der nicht-natürlichen Rückgratstruktur sind die Ergebnisse der Vorhersage der Seitenkettenstruktur jedes Algorithmus für die CASP13- und CASP14-Zielproteine. Sternchen zeigen an, dass die durchschnittlichen Konfliktwerte niedriger sind als die entsprechenden nativen Strukturen – 34,6, 2,2, 0,5 für CASP13 und 40,0, 2,7, 0,7 für CASP14.
Verzichtet innovativ auf diskrete Rotamer-Bibliotheken und rechenintensive Konformationssuch- und Probenahmeschritte und kombiniert direkt die geometrische 3D-Struktur der Hauptkette, um alle Seitenkettenkoordinaten parallel zu berechnen. Im Vergleich zu der auf Deep Learning basierenden Methode DLPacker und der auf traditionellen Computermethoden basierenden RosettaPacker hat AttnPacker die Recheneffizienz erheblich verbessert und die Inferenzzeit um mehr als das Hundertfache verkürzt.

Zeitvergleich verschiedener PSCP-Methoden. Rekonstruktion der relativen Zeiten der Seitenkettenatome für alle 83 CASP13-Zielproteine.
AttnPacker schneidet beim Proteindesign ebenso gut ab. Molecular Heart hat eine AttnPacker-Variante für das Co-Design trainiert, die native Sequenzwiederherstellungsraten erreicht, die mit aktuellen Methoden auf dem neuesten Stand der Technik vergleichbar sind, und gleichzeitig hochpräzise Baugruppen produziert. Die Validierung der Rosetta-Simulation zeigt, dass von AttnPacker entworfene Strukturen im Allgemeinen subnative (niedrigere) Rosetta-Energien erzeugen.
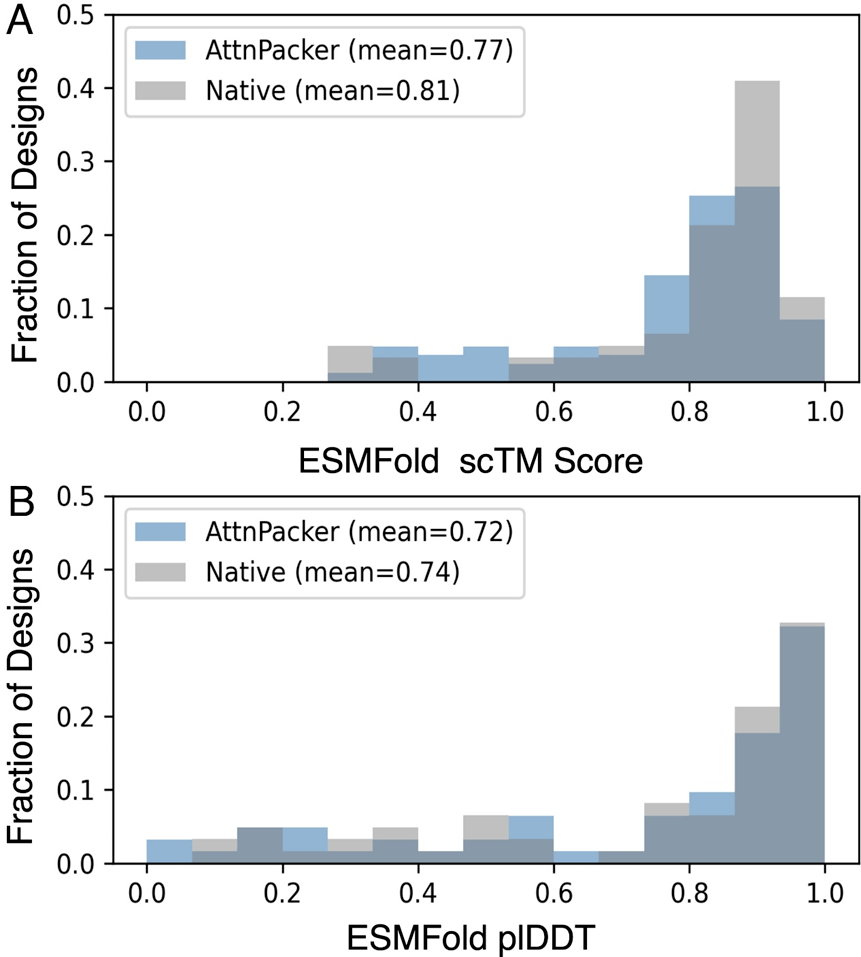
Unter Verwendung der ESMFold scTM- und plDDT-Indikatoren zum Vergleich der nativen Proteinsequenz und der von AttnPacker generierten Sequenz zur Bewertung der Qualität der AttnPacker-Generierung zeigten die Ergebnisse eine starke Korrelation.
Zusätzlich zu seiner erstaunlichen Effektivität und Effizienz hat AttnPaker auch einen sehr praktischen Wert – es ist sehr einfach zu bedienen. Für die Ausführung von AttnPaker ist lediglich eine Proteinstrukturdatei erforderlich. Im Gegensatz dazu erfordert OPUS-Rota4 (28) eine Voxeldarstellung der atomaren Umgebung von DLPacker, Logik, Sekundärstruktur von trRosetta100 und Einschränkungsdateien von der OPUS-CM-Ausgabe. Da AttnPacker außerdem Seitenkettenkoordinaten direkt vorhersagt, ist die Ausgabe vollständig differenzierbar, was nachgelagerte Vorhersageaufgaben wie Optimierung oder Protein-Protein-Wechselwirkungen erleichtert. „Die Vorteile des guten Vorhersageeffekts, der hohen Effizienz und der Benutzerfreundlichkeit begünstigen den weit verbreiteten Einsatz von AttnPacker in Forschungs- und Industriebereichen“, sagte Professor Xu Jinbo.
Zusammenfassung
1. AttnPacker ist ein äquivalentes SE (3)-Modell, das zur direkten Vorhersage der Proteinseitenkettenstruktur und des Proteinsequenzdesigns verwendet werden kann.
2. Die Genauigkeit von AttnPacker ist besser als bei anderen Methoden, die Effizienz ist erheblich verbessert und die Verwendung ist äußerst einfach.
Das obige ist der detaillierte Inhalt vonWeltneuheit: Der neue KI-Algorithmus von Molecular Heart zur Überwindung der Probleme der Proteinseitenkettenvorhersage und des Sequenzdesigns. Für weitere Informationen folgen Sie bitte anderen verwandten Artikeln auf der PHP chinesischen Website!
In Verbindung stehende Artikel
Mehr sehen- Technologietrends, die Sie im Jahr 2023 im Auge behalten sollten
- Wie künstliche Intelligenz Rechenzentrumsteams neue Alltagsaufgaben beschert
- Können künstliche Intelligenz oder Automatisierung das Problem der geringen Energieeffizienz in Gebäuden lösen?
- OpenAI-Mitbegründer im Interview mit Huang Renxun: Die Argumentationsfähigkeiten von GPT-4 haben noch nicht die Erwartungen erfüllt
- Dank der OpenAI-Technologie übertrifft Bing von Microsoft Google im Suchverkehr