
Heim >Technologie-Peripheriegeräte >KI >Erforschung und Anwendung von Modellen zur Vorhersage der Genomkonformation und rechnergestützten genetischen Screening-Methoden mit hohem Durchsatz
Erforschung und Anwendung von Modellen zur Vorhersage der Genomkonformation und rechnergestützten genetischen Screening-Methoden mit hohem Durchsatz

- PHPznach vorne
- 2023-05-08 14:16:08897Durchsuche

Abbildung 0
Unterschiede in der Genomkonformation in verschiedenen Zelltypen bestimmen die Spezifität der Genexpression, die wiederum die funktionellen Unterschiede in verschiedenen Zelltypen bestimmt. Lange Zeit waren experimentelle Methoden zur Genomkonformationsdetektion, von der In-situ-Hybridisierung bis zur Hochdurchsatzdetektion wie Hi-C- und Micro-C-Technologien, in der Regel zeitaufwändig, arbeitsintensiv, kostspielig und mit starken technischen Einschränkungen verbunden. Diese Methoden schränken die weit verbreitete Anwendung dieser experimentellen Techniken im Bereich der Genomkonformationsforschung erheblich ein, insbesondere bei der Untersuchung seltener Zelltypen und der Notwendigkeit, den kausalen Zusammenhang der Genomkonformationsregulation in großem Maßstab zu überprüfen. Die Einschränkungen dieser Methoden haben auch neue Entdeckungen auf dem Gebiet der dreidimensionalen Genomkonformationsregulation lange Zeit eingeschränkt.

Abbildung 1
9. Januar 2023, Aristotelis Tsirigos Laboratory und Broad Institute of MIT und Harvard, NYU Grossman School of Medicine Xia Bos Labor hat zusammengearbeitet, um einen Artikel „Zelltypspezifisch“ zu veröffentlichen „Vorhersage der 3D-Chromatinorganisation ermöglicht Hochdurchsatz-in-silico-Genscreening“ in Nature Biotechnology.

Papieradresse: https://www.nature.com/articles/s41587-022-01612-8
In dieser Studie Ph.D., New York University School Sheng Tan Jimin und Dr auf dem Prinzip des genetischen Screenings. , ISGS)-Methode, die zur Identifizierung zelltypspezifischer funktioneller genomischer Elemente und zur Entdeckung neuer Mechanismen der Chromatin-Konformationsregulation verwendet wird.
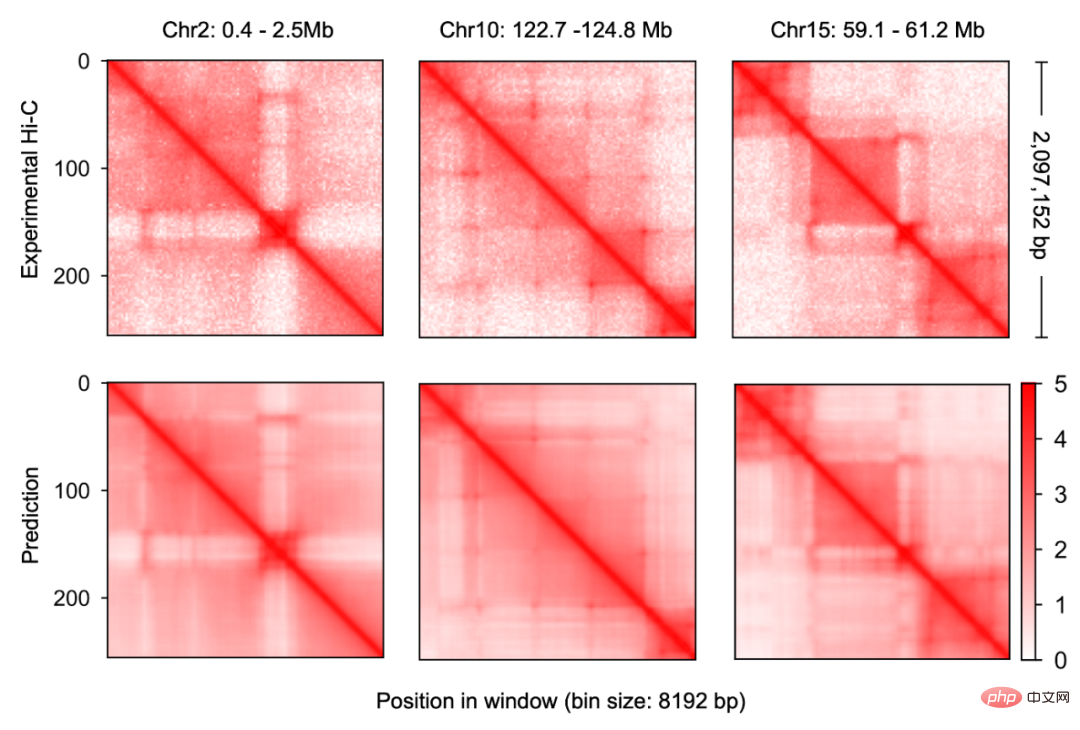
Abbildung 2
Forscher haben zunächst ein neues multimodales Deep-Learning-Framework für Genomdaten, Origami, entwickelt, damit DNA-Sequenzinformationen und zellspezifische funktionelle Genomdaten effektiv integriert werden können Informationen und prognostizieren dann neue genomische Informationen. Durch wiederholtes Debuggen und Modelltraining fanden die Forscher heraus, dass die Integration der DNA-Sequenz, des CTCF-Bindungsstatus (CTCF ChIP-seq) und der ATAC-seq-Signale als Eingabeinformationen die Chromatinkonformation genau vorhersagen kann und die zweidimensionale Hi-C-Matrix als verwendet Sagen Sie das Ausgabeziel voraus (Abbildung 1-2). Die Eingabeinformationen umfassten 2 Millionen Basenpaare DNA, CTCF ChIP-seq und ATAC-seq. Forscher verwenden Onehot-Kodierung, um diskrete DNA-Sequenzen zu kodieren, während CTCF ChIP-seq und ATAC-seq nicht-diskrete Merkmale kodieren.

C.Origami-Modell ist in drei Teile unterteilt: den Encoder, der DNA- und Genominformationen verarbeitet und komprimiert, die Transformer-Mittelschicht und den Ausgabe-Hi-C-Decoder . Der Encoder besteht aus einer Reihe von 1D-ResNet und schrittweiser Faltung, um die Eingabeinformationen von 2 Millionen Basenpaaren zu kodieren und zu komprimieren. Am Ende des Encoders wird die Nachricht mit einer Länge von 2 Millionen auf eine Länge von 256 komprimiert und als Eingabenachricht für den Transformer verwendet. Der Selbstaufmerksamkeitsmechanismus von Transformer kann die gegenseitige Abhängigkeit zwischen verschiedenen Genomregionen bewältigen und die Gesamtleistung des Modells verbessern. Die Aufmerksamkeitsmatrix in Transformer kann auch die Interpretierbarkeit des Modells verbessern. Die Forscher wandelten das Aufmerksamkeitsgewicht in einen „Aufmerksamkeitswert“ um, um die Betonung verschiedener Bereiche des Modells bei der Vorhersage zu messen. Schließlich wandelten die Forscher die 1D-Ausgabe des Transformer-Moduls mithilfe der „äußeren Verkettung“ in eine 2D-Kontakt-/Adjazenzmatrix um, die als Eingabeinformationen für den Hi-C-Decoder verwendet wurde. Der Decoder ist ein Dilated 2D ResNet. Die Forscher passten die Dilatationsfaktoren verschiedener Schichten an, sodass das Empfangsfeld an jeder Pixelposition der letzten Schicht alle Eingabeinformationen abdecken kann.
Dieses Modell zur Vorhersage der Chromatinkonformation heißt C.Origami. Forscher nennen C.Origami das erste multimodale Deep-Learning-Modell in der Genomik. Aufgrund seiner multimodalen Natur ist C.Origami in der Lage, die Chromatinkonformation in neuen Zelltypen, die noch nie zuvor exponiert wurden, genau vorherzusagen (De-novo-Vorhersage). Beispielsweise war ein auf IMR-90-Zellen (Lungenfibroblasten) trainiertes Modell in der Lage, spezifische Chromatinkonformationen in GM12878-Zellen (B-Lymphozyten) genau vorherzusagen (Abbildung 3).

Abbildung 3
Strukturelle Varianten – wie chromosomale Translokationen – kommen in Tumoren sehr häufig vor und verändern häufig Chromatin-Interaktionsmuster, was die Expression von Onkogenen oder Tumorsuppressorgenen beeinflussen kann. Die Untersuchung der Auswirkungen dieser strukturellen Variationen auf die Chromatinkonformation und Genexpression ist wichtig für das Verständnis der Mechanismen des Auftretens und Fortschreitens von Tumoren. Diese Art von Forschung erfordert normalerweise den Einsatz von Experimenten wie 4C-seq oder Hi-C, um die Chromatinkonformation struktureller Variationsstellen zu analysieren, ist jedoch oft durch Ressourcen und Zeit begrenzt und in großem Maßstab schwer durchzuführen.
In dieser Studie kann C.Origami DNA-Sequenzvariationen in Eingabevariablen simulieren und dann neue Chromatin-Wechselwirkungen im mutierten Krebsgenom vorhersagen. Frühere Studien ergaben, dass das Zellmodell CUTLL1 der akuten lymphatischen T-Zell-Leukämie (T-ALL) eine chromosomale chr7-chr9-Translokation aufweist (Abbildung 4). Durch die rechnerische Simulation chromosomaler Translokationsvarianten hat C. Origami die neue TAD-Struktur an der Variantenstelle genau vorhergesagt und eine „Chromatinstreifen“-Struktur entdeckt, die sich von chr9 bis chr7 erstreckt (Abbildung 4).

Abbildung 4
Angesichts der genauen Vorhersagewirkung von C.Origami und inspiriert vom Prinzip des umgekehrten genetischen Screenings schlugen die Forscher ein neues rechnergestütztes genetisches Screening mit hohem Durchsatz (in silico) vor genetisches Screening (ISGS) zur systematischen Identifizierung zelltypspezifischer funktioneller genomischer Elemente und zur Entdeckung neuer färbender regulatorischer Moleküle (Abbildung 5). Die Forscher entwickelten einen Rahmen für das computergestützte genetische Screening ISGS basierend auf dem C. Origami-Modell zur systematischen Identifizierung von cis-regulatorischen Elementen, die für die Chromatinkonformation erforderlich sind. Mithilfe der genomweiten ISGS mit 1 kb Auflösung isolierten die Autoren cis-regulatorische Elemente (ca. 1 % des Genoms), die wichtige Auswirkungen auf die Chromatinkonformation haben. Diese regulatorischen Sequenzen der Chromatinkonformation weisen eine unterschiedliche Abhängigkeit von der CTCF-Bindung und den ATAC-seq-Signalen auf (Abb. 5).

Bild 5
ISGS-Framework ermöglicht Hochdurchsatz-Screening zell- oder krankheitsspezifischer Chromatinkonformationen. Die Forscher führten ISGS in CUTLL1-, Jurkat- und normalen T-Zellen durch und stellten fest, dass ein cis-regulatorisches Element (CHD4-insu) in der Nähe des CHD4-Gens spezifisch in T-ALL-Zellen verloren ging. Screening-Ergebnisse deuten darauf hin, dass der isolierende Verlust von CHD4-insu in T-ALL-Zellen es dem CHD4-Gen ermöglichen könnte, neue Chromatin-Interaktionen aufzubauen, wodurch die CHD4-Expression hochreguliert und die Proliferation von Leukämiezellen gefördert wird.
ISGS kann auch zur systematischen Entdeckung neuer transaktiver Faktoren verwendet werden, die die Chromatinkonformation regulieren. Durch die Anreicherungsanalyse wichtiger zelltypspezifischer regulatorischer Sequenzen und Transkriptionsfaktor-Bindungsstellen identifizierten die Forscher regulatorische Faktoren, die zur zelltypspezifischen Genomkonformation beitragen. Interessanterweise ergaben frühere Studien, dass MAZ möglicherweise zusammen mit CTCF die Chromatinkonformation reguliert. Durch ISGS- und Transkriptionsfaktor-Anreicherungsanalyse stellten die Autoren fest, dass MAZ in offenen Chromatinregionen stark angereichert ist, während es in nichtoffenen Chromatinregionen, an denen CTCF bindet, nur eine schwache Bindung zeigt. Dieses Ergebnis legt nahe, dass MAZ die Genomkonformation unabhängig von CTCF regulieren könnte.
Forscher sehen großes Potenzial in multimodalen Modellen des maschinellen Lernens, die DNA-Sequenz- und Chromatininformationen zur Vorhersage der Chromatinstruktur kombinieren. Die zugrunde liegende multimodale Architektur des Modells, Origami, kann auf die Anwendung anderer genomischer Daten erweitert werden, wie etwa epigenetische Modifikationen, Genexpression, funktionelles Screening von Mutationen usw. Forscher gehen davon aus, dass sich die Genomforschung in Zukunft stärker auf den Einsatz von Deep-Learning-Modellen als Werkzeugen für das primäre computergestützte genetische Screening konzentrieren wird, ergänzt durch eine neue Generation von Hochdurchsatz-Forschungsmethoden, die durch biologische Experimente validiert werden.
In dieser Studie ist Tan Jimin, Doktorand an der New York University School of Medicine, der Erstautor, und Dr. Aristotelis Tsirigos und Dr. Xia Bo sind die Mitautoren. Diese Forschung begann mit dem Brainstorming von Xia Bo und Tan Jimin während des epidemischen Lockdowns im Oktober 2020. Nach zweieinhalb Jahren der Verbesserung und Ausarbeitung wurde sie im Januar 2023 offiziell in Nature Biotechnology veröffentlicht.
Der Code und die Trainingsdaten dieses Projekts wurden als Open Source auf GitHub und Zenodo bereitgestellt und sind zur Funktionsdemonstration mit Google Colab ausgestattet.
Projektadresse: https://github.com/tanjimin/C.Origami
Korrespondierender Autor
Dr. Xia Bo’s Laboratory (Broad Institute of MIT und Harvard) Homepage: www.boxialab.org
Dr. Xia Bo widmet sich der Analyse des Kernmechanismus zur Regulierung der dreidimensionalen Konformation des Genoms und seiner biologischen Bedeutung für menschliche Krankheiten, Entwicklung und Evolution. Das Labor von Xia Bo heißt gleichgesinnte Postdoktoranden willkommen, die dem Team beitreten möchten.
Tsirigos Lab (New York University Grossman School of Medicine) Homepage: http://www.tsirigos.com
Zu den Hauptforschungsrichtungen des Tsirigos Lab gehören Chromatin, Epigenetik und maschinelles Lernen in Anwendungen der Präzisionsmedizin.
Das obige ist der detaillierte Inhalt vonErforschung und Anwendung von Modellen zur Vorhersage der Genomkonformation und rechnergestützten genetischen Screening-Methoden mit hohem Durchsatz. Für weitere Informationen folgen Sie bitte anderen verwandten Artikeln auf der PHP chinesischen Website!
In Verbindung stehende Artikel
Mehr sehen- Technologietrends, die Sie im Jahr 2023 im Auge behalten sollten
- Wie künstliche Intelligenz Rechenzentrumsteams neue Alltagsaufgaben beschert
- Können künstliche Intelligenz oder Automatisierung das Problem der geringen Energieeffizienz in Gebäuden lösen?
- OpenAI-Mitbegründer im Interview mit Huang Renxun: Die Argumentationsfähigkeiten von GPT-4 haben noch nicht die Erwartungen erfüllt
- Dank der OpenAI-Technologie übertrifft Bing von Microsoft Google im Suchverkehr