
Heim >Technologie-Peripheriegeräte >KI >Meta AI öffnet mehr als 600 Millionen metagenomische Proteinstrukturkarten und 15 Milliarden Sprachmodelle wurden in zwei Wochen fertiggestellt
Meta AI öffnet mehr als 600 Millionen metagenomische Proteinstrukturkarten und 15 Milliarden Sprachmodelle wurden in zwei Wochen fertiggestellt

- 王林nach vorne
- 2023-04-16 11:37:021830Durchsuche
In diesem Jahr veröffentlichte DeepMind die vorhergesagten Strukturen von etwa 220 Millionen Proteinen, was fast alle Proteine bekannter Organismen in der DNA-Datenbank abdeckt. Jetzt füllt ein weiterer Technologieriese, Meta, eine weitere Lücke, nämlich die der Mikroben.
Einfach ausgedrückt: Meta nutzt KI-Technologie, um die Strukturen von etwa 600 Millionen Proteinen aus Bakterien und anderen noch zu charakterisierenden Mikroorganismen vorherzusagen. Teamleiter Alexander Rives sagte: „Diese Proteine sind die Strukturen, über die wir am wenigsten wissen, und es sind sehr mysteriöse Proteine. Ich denke, diese Erkenntnisse bieten das Potenzial für ein tiefes Verständnis der Biologie.“ große Mengen Auf Text geschult. Meta Um Sprachmodelle auf Proteine anzuwenden, verwendeten Rives und Kollegen bekannte Proteinsequenzen als Eingabe, die aus 20 Aminosäuren bestehen, die durch verschiedene Buchstaben dargestellt werden. Das Netzwerk lernte dann, Proteine automatisch zu vervollständigen und gleichzeitig einen bestimmten Anteil an Aminosäuren zu maskieren.
Meta hat dieses Netzwerk ESMFold genannt. Obwohl die Vorhersagegenauigkeit von ESMFold nicht so gut ist wie die von AlphaFold, ist es bei der Vorhersage von Strukturen etwa 60-mal schneller als AlphaFold. Diese Geschwindigkeit bedeutet, dass Vorhersagen der Proteinstruktur auf größere Datenbanken übertragen werden können.

- project Adresse: https://github.com/facebookresearch/esm
- Als Test entschied sich Meta nun, ihr Modell auf eine Datenbank metagenomischer DNA anzuwenden, die alle aus der Umwelt stammt, einschließlich Erde, Meerwasser, menschlichem Darm, Haut und anderen mikrobiellen Lebensräumen. Meta AI
. Es ist außerdem die größte Datenbank mit hochauflösenden vorhergesagten Strukturen, dreimal größer als jede bestehende Proteinstrukturdatenbank, und die erste, die eine umfassende, groß angelegte Abdeckung metagenomischer Proteine bietet.
Insgesamt hat das Meta-Team in nur zwei Wochen mehr als 617 Millionen Proteinstrukturen vorhergesagt. Rives sagte, die Vorhersagen seien kostenlos und für jedermann verfügbar, genau wie der zugrunde liegende Code des Modells. 
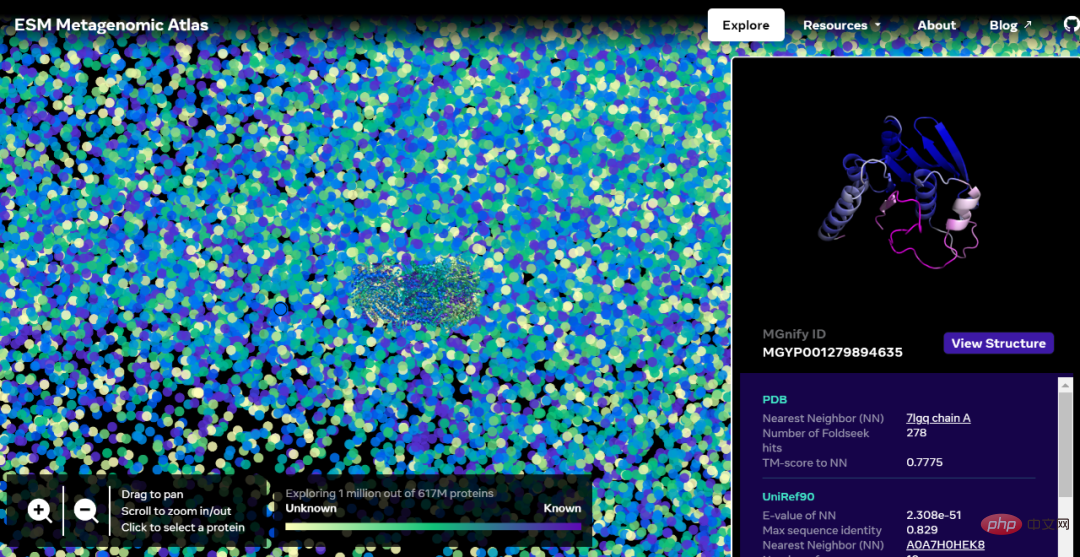 Adresse der interaktiven Version: https://esmatlas.com/explore?at=1%2C1%2C21.999999344348925
Adresse der interaktiven Version: https://esmatlas.com/explore?at=1%2C1%2C21.999999344348925
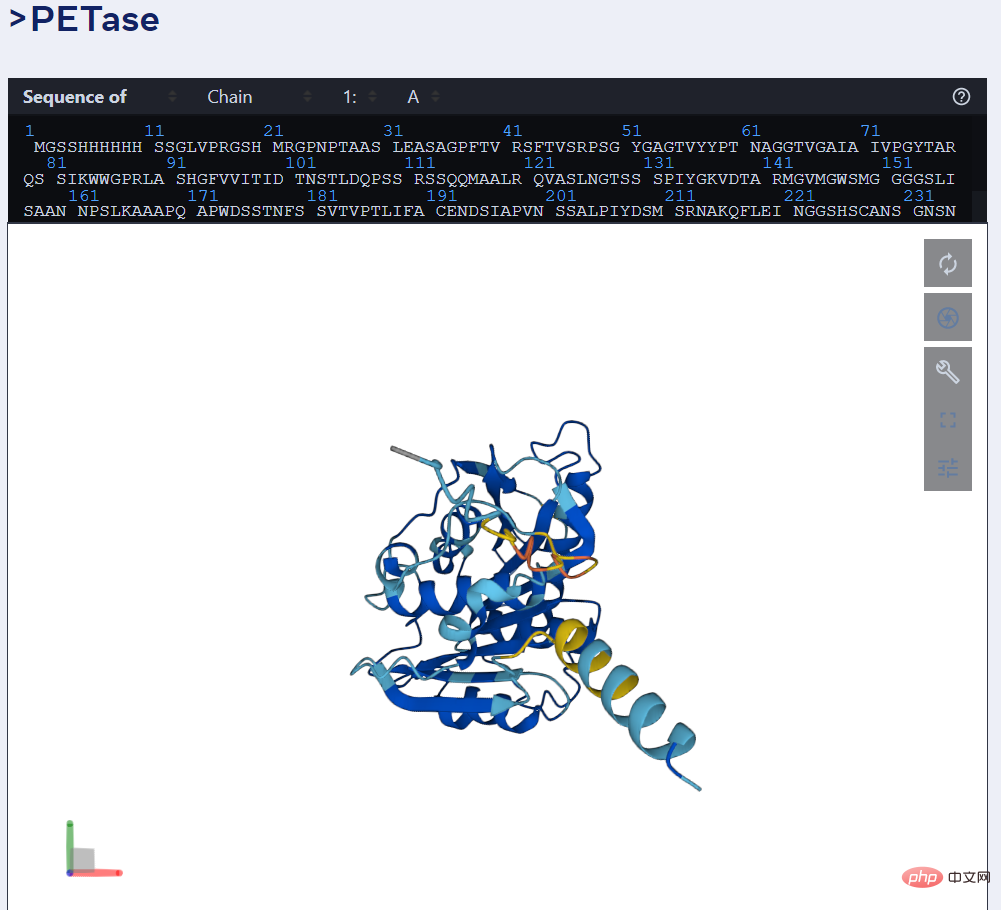
Das Bild unten zeigt beispielsweise ESMFolds Vorhersage des PET-Enzyms.
Einführung
Im Jahr 1998 schlug Jo Handelsman von der Abteilung für Pflanzenpathologie der University of Wisconsin erstmals das Konzept der Metagenomik vor, das aus der Untersuchung und Analyse von Gensätzen aus der Umwelt in gewisser Weise als einzelnes Genom entstand of und Makro im Englischen ist meta-, was auch als Yuan übersetzt wird.
Metagenomics enthüllt Milliarden von Proteinsequenzen, die für die Wissenschaft neu sind und zum ersten Mal vom NCBI (European Bioinformatics Institute und Joint Genome Institute) und anderen großen Datenbanken, die im Rahmen öffentlicher Projekte zusammengestellt wurden, katalogisiert wurden.
Meta AI hat eine neue Proteinfaltungsmethode entwickelt, die große Sprachmodelle nutzt, um die erste umfassende Ansicht der Proteinstruktur in metagenomischen Datenbanken (mit Hunderten Millionen Proteinen) zu erstellen. Meta fand heraus, dass Sprachmodelle die dreidimensionale Struktur von Proteinen auf atomarer Ebene 60-mal schneller vorhersagen können als bestehende SOTA-Methoden zur Vorhersage der Proteinstruktur. Dieser Fortschritt wird dazu beitragen, eine neue Ära des Proteinstrukturverständnisses zu beschleunigen und es erstmals möglich zu machen, die Strukturen der Milliarden von Proteinen zu verstehen, die durch genetische Sequenzierungstechnologie katalogisiert werden.
Erschließung der verborgenen Welt der Natur: der erste umfassende Blick auf den metagenomischen Strukturraum
Wir wissen, dass Fortschritte in der genetischen Sequenzierung die Analyse ermöglichen Die Katalogisierung von Milliarden metagenomischer Proteinsequenzen wurde möglich. Die experimentelle Bestimmung der 3D-Struktur von Milliarden von Proteinen geht jedoch weit über den Rahmen zeitintensiver Labortechniken wie der Röntgenkristallographie hinaus, deren Nachweis eines einzelnen Proteins Wochen oder sogar Jahre dauern kann. Computergestützte Ansätze können Einblicke in metagenomische Proteine liefern, die mit experimentellen Techniken nicht möglich sind.
Der ESM Metagenomic Atlas wird es Wissenschaftlern ermöglichen, die Struktur metagenomischer Proteine auf einer Skala von Hunderten Millionen Proteinen zu suchen und zu analysieren. Dies kann dabei helfen, bisher nicht charakterisierte Strukturen zu identifizieren, nach entfernten evolutionären Beziehungen zu suchen und neue Proteine zu entdecken, die in der Medizin und anderen Anwendungen verwendet werden könnten.
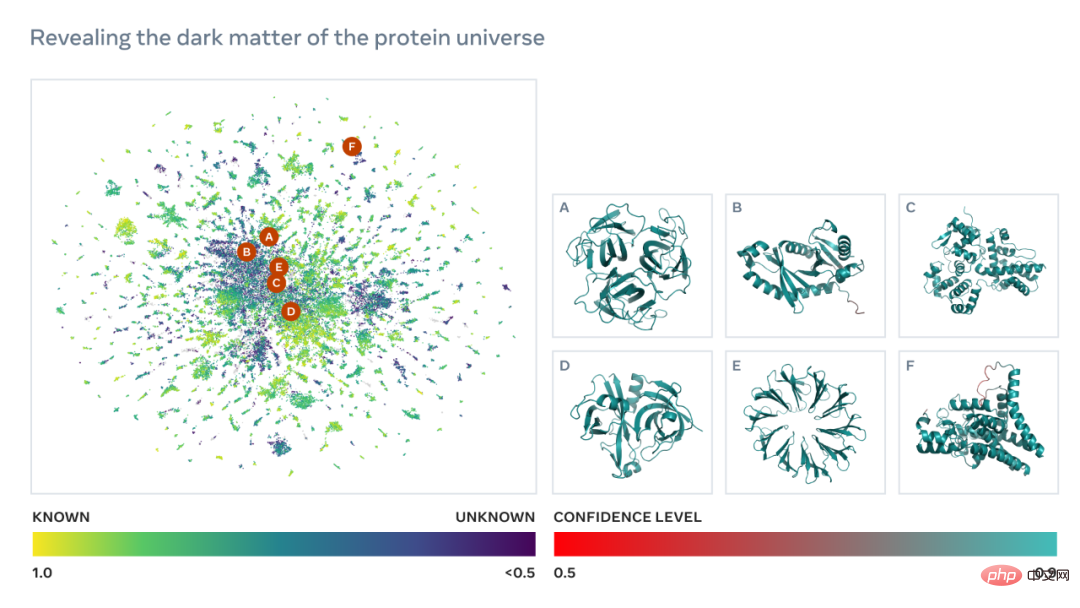
Nachfolgend finden Sie eine Karte mit Zehntausenden hochzuverlässigen Vorhersagen, die Ähnlichkeiten zu Proteinen mit derzeit bekannten Strukturen zeigen. Und zum ersten Mal zeigt das Bild einen viel größeren Bereich des Proteinstrukturraums, der völlig unbekannt war.
Biologiesprache lesen lernen
Wie unten gezeigt, ESM-2-Sprache Das Modell wird darauf trainiert, Aminosäuren vorherzusagen, die während der Evolution durch Sequenzen maskiert wurden. Meta AI stellte fest, dass als Ergebnis des Trainings Informationen über die Proteinstruktur im internen Zustand des Modells auftauchten. Dies ist überraschend, da das Modell nur auf Sequenzen trainiert wurde. Wie der Text einer Arbeit oder eines Briefes können Proteine als Zeichenfolge geschrieben werden. Jedes Zeichen entspricht einem von 20 chemischen Standardelementen (Aminosäuren), die jeweils unterschiedliche Eigenschaften haben und die Bausteine von Proteinen sind. Diese Bausteine können auf astronomisch unterschiedliche Weise zusammengesetzt werden, zum Beispiel gibt es für ein Protein, das aus 200 Aminosäuren besteht, 20^200 mögliche Sequenzen, was mehr ist als die Anzahl der Atome im sichtbaren Universum. Jede Sequenz faltet sich in eine 3D-Form (aber nicht alle Sequenzen falten sich in eine kohärente Struktur, viele falten sich in eine ungeordnete Form), und diese Form bestimmt weitgehend die biologische Funktion des Proteins.
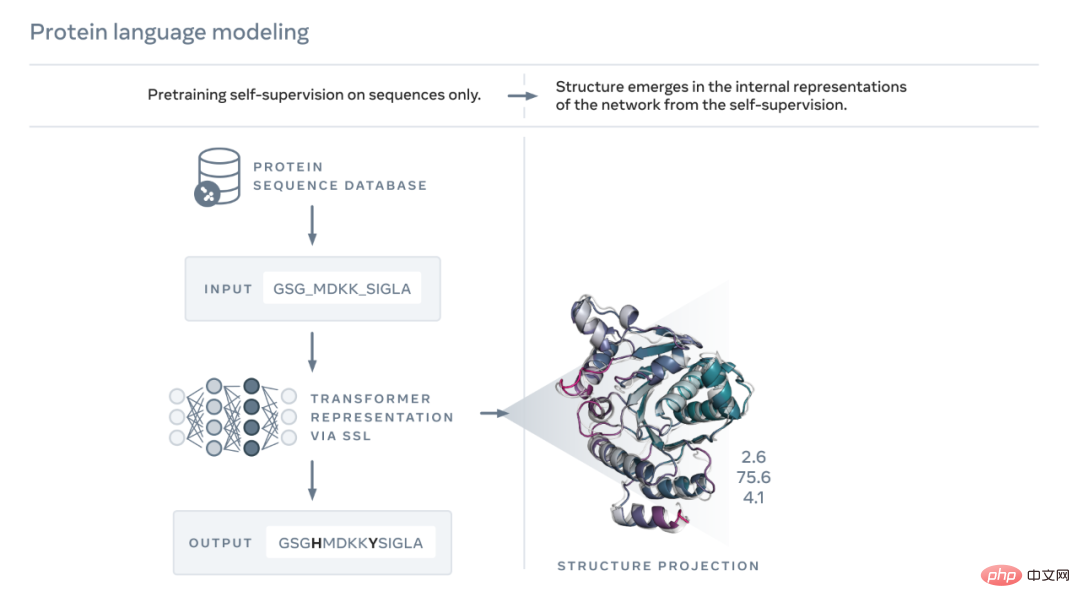 Das Erlernen des Lesens der biologischen Sprache bringt große Herausforderungen mit sich. Obwohl sowohl Proteinsequenzen als auch Textpassagen als Zeichen geschrieben werden können, gibt es zwischen ihnen tiefe und grundlegende Unterschiede. Eine Proteinsequenz beschreibt die chemische Struktur eines Moleküls, das sich gemäß den Gesetzen der Physik in komplexe 3D-Formen faltet.
Das Erlernen des Lesens der biologischen Sprache bringt große Herausforderungen mit sich. Obwohl sowohl Proteinsequenzen als auch Textpassagen als Zeichen geschrieben werden können, gibt es zwischen ihnen tiefe und grundlegende Unterschiede. Eine Proteinsequenz beschreibt die chemische Struktur eines Moleküls, das sich gemäß den Gesetzen der Physik in komplexe 3D-Formen faltet.
Proteinsequenzen enthalten statistische Muster, die Informationen über die Proteinfaltungsstruktur vermitteln. Wenn sich beispielsweise zwei Positionen in einem Protein gemeinsam entwickeln, oder mit anderen Worten, wenn eine bestimmte Aminosäure an einer Position vorkommt, die normalerweise mit einer bestimmten Aminosäure an der anderen Position gepaart ist, kann dies bedeuten, dass sich die beiden Positionen an der gleichen Stelle befinden Interaktion der gefalteten Struktur. Dies ähnelt zwei Teilen eines Puzzles, bei dem die Evolution die Aminosäuren auswählen muss, die in einer gefalteten Struktur zusammenpassen. Das wiederum bedeutet, dass wir oft aus der Beobachtung von Mustern in seiner Sequenz auf die Struktur eines Proteins schließen können.
ESM nutzt KI, um zu lernen, diese Muster zu lesen. Im Jahr 2019 lieferte Meta AI den Beweis, dass Sprachmodelle die Eigenschaften von Proteinen, etwa deren Struktur und Funktion, lernen. Durch eine Form des selbstüberwachten Lernens namens Masked Language Modeling trainierte Meta AI ein Sprachmodell auf den Sequenzen von Millionen natürlicher Proteine. Mit dieser Methode muss das Modell die Lücken im Textabsatz korrekt ausfüllen, z. B. „To _ or not to , that is the _____“.
Anschließend trainiert Meta AI ein Sprachmodell, um die Lücken in der Proteinsequenz zu füllen. Sie fanden heraus, dass während dieses Trainings Informationen über die Struktur und Funktion von Proteinen gewonnen wurden. Im Jahr 2020 veröffentlichte Meta ein SOTA-Proteinsprachenmodell, ESM1b, für eine Vielzahl von Anwendungen, darunter die Unterstützung von Wissenschaftlern bei der Vorhersage der Entwicklung von COVID-19 und der Entdeckung der genetischen Ursachen der Krankheit.
Jetzt hat Meta AI diesen Ansatz erweitert, um das Protein-Sprachmodell der nächsten Generation ESM-2 zu erstellen, das mit 15 Milliarden Parametern das bislang größte Protein-Sprachmodell ist. Sie fanden heraus, dass bei einer Skalierung der Modellparameter von 8 Millionen auf 15 Milliarden Informationen in der internen Darstellung auftauchten, was 3D-Strukturvorhersagen mit atomarer Auflösung ermöglichte.
Erzielen Sie eine Beschleunigung der Proteinfaltung um Größenordnungen
In der Abbildung unten erscheint beim Vergrößern des Modells eine hochauflösende Proteinstruktur. Gleichzeitig erscheinen bei der Skalierung des Modells neue Details in atomar aufgelösten Bildern der Proteinstruktur.

Mit den aktuellen SOTA-Rechentools könnte die Vorhersage der Struktur von Hunderten Millionen Proteinsequenzen in einem realistischen Zeitrahmen Jahre dauern, selbst mit den Ressourcen großer Forschungseinrichtungen. Um Vorhersagen im metagenomischen Maßstab treffen zu können, ist daher ein Durchbruch in der Vorhersagegeschwindigkeit von entscheidender Bedeutung.
Meta AI hat herausgefunden, dass die Verwendung von Sprachmodellen von Proteinsequenzen die Strukturvorhersage erheblich beschleunigt, und zwar um das bis zu 60-fache. Dies reicht aus, um in nur wenigen Wochen Vorhersagen für eine gesamte metagenomische Datenbank zu treffen, und kann auf viel größere Datenbanken als unsere derzeit veröffentlichten Datenbanken skaliert werden. Tatsächlich war diese neue Strukturvorhersagefunktion in der Lage, die Sequenzen von mehr als 600 Millionen Metagenomproteinen in nur zwei Wochen auf einem Cluster von etwa 2.000 GPUs vorherzusagen.
Darüber hinaus erfordern aktuelle SOTA-Strukturvorhersagemethoden die Suche in großen Proteindatenbanken, um relevante Sequenzen zu identifizieren. Diese Methoden erfordern im Wesentlichen einen ganzen Satz evolutionär verwandter Sequenzen als Eingabe, damit sie strukturbezogene Muster extrahieren können. Das ESM-2-Sprachmodell von Meta AI lernt diese Evolutionsmuster während des Trainings an Proteinsequenzen und ermöglicht so hochauflösende Vorhersagen von 3D-Strukturen direkt aus Proteinsequenzen.
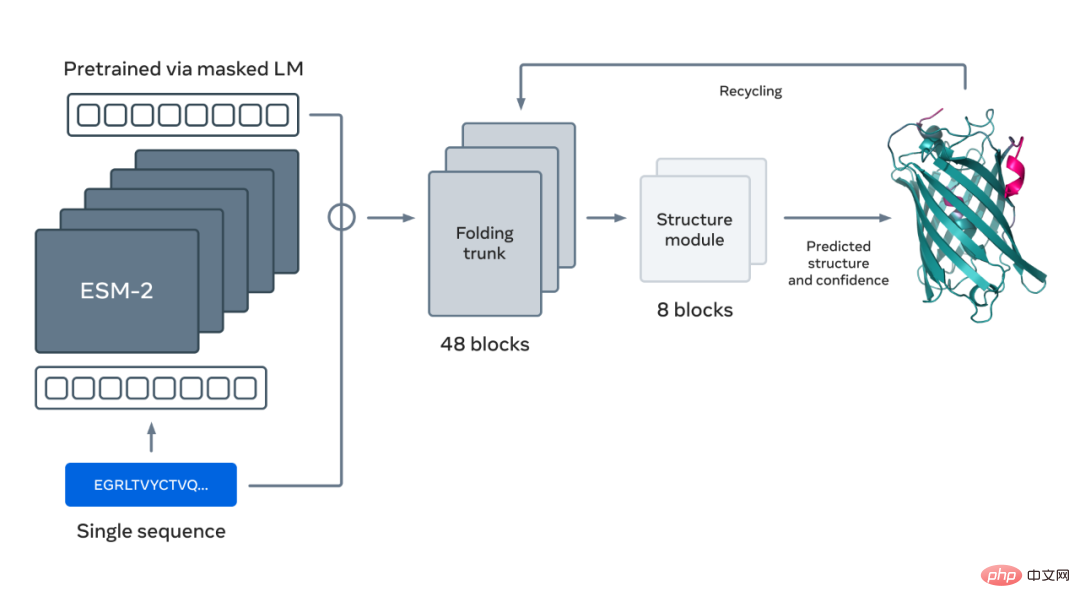
Die Abbildung unten zeigt die Proteinfaltung mit dem ESM-2-Sprachmodell. Die Pfeile von links nach rechts zeigen den Informationsfluss im Netzwerk vom Sprachmodell über den Faltstamm bis zum Strukturmodul und geben schließlich 3D-Koordinaten und Konfidenz aus.
Weitere Informationen finden Sie im Originalartikel.
Blog-Link: https://ai.facebook.com/blog/protein-folding-esmfold-metagenomics/
Das obige ist der detaillierte Inhalt vonMeta AI öffnet mehr als 600 Millionen metagenomische Proteinstrukturkarten und 15 Milliarden Sprachmodelle wurden in zwei Wochen fertiggestellt. Für weitere Informationen folgen Sie bitte anderen verwandten Artikeln auf der PHP chinesischen Website!
In Verbindung stehende Artikel
Mehr sehen- Technologietrends, die Sie im Jahr 2023 im Auge behalten sollten
- Wie künstliche Intelligenz Rechenzentrumsteams neue Alltagsaufgaben beschert
- Können künstliche Intelligenz oder Automatisierung das Problem der geringen Energieeffizienz in Gebäuden lösen?
- OpenAI-Mitbegründer im Interview mit Huang Renxun: Die Argumentationsfähigkeiten von GPT-4 haben noch nicht die Erwartungen erfüllt
- Dank der OpenAI-Technologie übertrifft Bing von Microsoft Google im Suchverkehr