
计算效率提升100倍以上,上交李金金团队开发基于Transformer的大模型用于从头算分子动力学

- WBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWB原创
- 2024-06-18 18:02:541254浏览

作者| 陶科豪
精确模拟原子与分子的动态行为对于开发新一代高效能材料至关重要。
然而,传统的从头算分子动力学(AIMD)模拟自然提供了高精度的预测能力,但由于其高昂的计算成本和漫长的模拟时间,大大限制了研究的进度。
比如,建立一个包含100个原子的材料系统的30皮秒模拟通常需要数月时间,这对于需要快速迭代和优化的新材料研发构成了巨大挑战。
在这种背景下,一个能够显着加快这一过程的人工智能模型具有重要价值。
面对这些挑战,上海交通大学人工智能与微结构实验室(AIMS-lab)开发了名为 T-AIMD 的革命性人工智能模型。
这种模型采用了基于Transformer网络架构,不仅能够高效降低计算成本,同时也能快速准确地预测任何离子在任何晶体结构中的行为。
通过这种方式,T-AIMD 模型将传统的 AIMD 模拟速度提升了 100 倍以上,显着加速了材料性能的评估过程。
此外,该模型还成功构建了一个庞大的混合离子导体数据库,并在多项电池实验中验证了其预测的准确性。
该方法不仅在分子动力学模型(MD),生物药物分子结合靶标、蛋白质折叠、材料热力学过程和力学性能计算等领域具有广泛的应用潜力。
也为使用生成式人工智能模型在更广泛的科学领域内解决复杂问题提供了新的方法论。
T-AIMD 的成功应用展示了人工智能技术在推动科学研究和技术创新中的巨大潜力,为未来的新材料研发和生物设计开发开辟了新的道路。
该研究以「Transformer enables ion transport behavior evolution and conductivity regulation for solid electrolyte」为题,于2024 年6 月11日发表在国际著名期刊《Energy Storage Materials》上。
论文的第一作者为上海交通大学人工智能与微结构实验室博士生陶科豪,通讯作者为实验室主任李金金教授。

文章链接:https://www.sciencedirect.com/science/article/pii/S2405829724003829
在人工智能领域,Transformer 模型因其卓越的并行处理能力和出色的性能,已经成为处理复杂序列数据的首选框架。
这种模型特别擅长从大规模数据中学习深层次的模式和关联,因此在语言处理、图像识别以及各类预测任务中得到了广泛应用。
尽管如此,在材料科学特别是从头算分子动力学(AIMD)模拟的应用中, Transformer 的潜力尚未得到充分开发。
传统的 AIMD 模拟在材料科学中非常重要,它能够精确模拟原子和分子的动态行为。然而,这类模拟通常依赖于重复的计算和昂贵的实验,不仅耗时而且成本高昂。
面对这样的挑战,一个能够快速提取和处理大量序列数据的智能模型显得尤为重要。
针对这一需求,上海交通大学 AIMS-lab 团队开发的 T-AIMD 模型,利用 Transformer 网络架构,显着提升了 AIMD 模拟的速度和准确性。
这种新型模型能够在极大降低计算成本的同时,快速准确地分析和预测原子及分子在各种条件下的行为。
与传统 AIMD 方法相比,T-AIMD 能将模拟速度提高 100 倍以上,同时保持了预测的高准确性,大幅缩短了材料研发的周期。
这不仅为材料科学领域的研究提供了新的工具,也展示了 AI 在高性能计算任务中的应用潜力,为未来的科学探索开辟了新的可能。
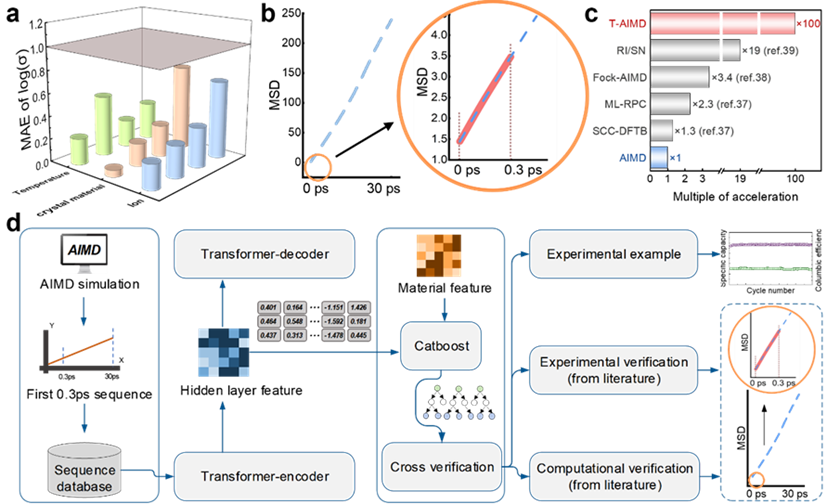
图示:T-AIMD 预测结果示意及工作流程示意图。 (来源:论文)
以解决固态电解质中离子输运行为的预测问题为例。该模型通过学习离子在电解质中的扩散序列,能够预测其在未来状态下的行为,极大地加速了材料性能的评估过程。
此外,T-AIMD 模型还结合了多源材料描述符,增强了其在处理复杂材料系统中的应用能力,使其不仅能预测单一离子种类的行为,还能处理多离子系统中的交互作用和复杂动力学问题。
这种基于 Transformer 的新方法为固态电解质的研发提供了一种全新的视角和工具,有望在材料科学领域开创新的研究和应用前景。
关于 T-AIMD 如何工作
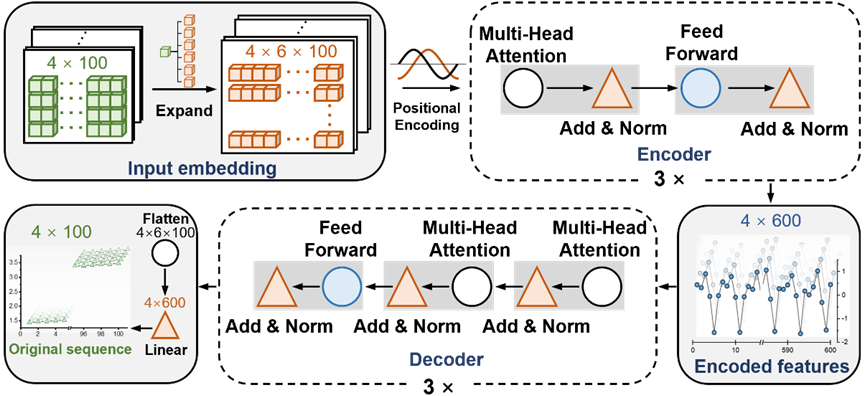
图示:T-AIMD 的网络架构图。(来源:论文)
T-AIMD(Transformer-based Ab Initio Molecular Dynamics)是一种结合了从头算分子动力学(AIMD)模拟和 Transformer 深度学习架构的模型,旨在提高固态电解质材料中离子输运特性的预测速度和准确性。这种模型的工作原理可以分为以下几个关键步骤:
1、 数据准备和预处理
T-AIMD 首先收集材料的离子扩散数据,这些数据来自于传统的AIMD模拟。这些模拟生成的数据包括时间序列数据,记录了离子在电解质中的移动轨迹。对这些序列数据进行预处理,将其转换为适合机器学习模型输入的格式。
2、 特征提取
利用 Transformer 模型的编码器部分,T-AIMD 能够从序列数据中提取关键特征。这一过程中,模型通过自注意力机制捕捉序列中的长距离依赖关系,这对于理解复杂的离子动力学非常关键。
3、 序列学习和预测
在特征提取后,Transformer 模型的解码器部分被用来基于已编码的特征进行序列预测。这一步骤中,模型不仅可以预测离子的未来行为,还可以分析在不同条件下(如不同温度和压力)离子的潜在行为。此外,模型通过这些学习到的特征,能够预测材料的离子导电性等关键性能指标。
4、多源材料描述符的集成
T-AIMD 结合了来自不同源的材料描述符,如晶体结构、离子种类和电子属性等,这有助于模型更全面地理解和预测材料性能。这种集成方法提高了模型在不同材料系统中的通用性和适应性。
5、 模型验证和应用
开发完成的模型需要通过与实验数据和其他计算方法的比较来验证其预测准确性。验证成功后,T-AIMD 可以用于快速筛选和优化新的目标材料,大大缩短研发周期并降低成本。
关于 T-AIMD 的稳健性能
T-AIMD 模型的稳健性能主要表现在以下几个方面:
1、准确性
T-AIMD 模型整合了 Transformer 架构,极大增强了其学习和预测复杂动力学行为的能力。在 AIMD 模拟加速方面,T-AIMD 显示出比传统方法更高的准确性。这得益于深度学习技术的应用,使模型能够在更短的时间内精确预测更长时间尺度的离子行为。
2、计算效率
在计算效率方面,T-AIMD 显著优于传统 AIMD 方法。传统的 AIMD 模拟需耗费大量时间模拟离子扩散,而 T-AIMD 通过优化计算流程,显著降低了对高性能计算资源的依赖,将模拟时间从几个月缩短至几天或几小时。
3、通用性和灵活性
T-AIMD 能处理比传统机器学习模型(如支持向量机或决策树)更复杂的数据结构和更大的数据集。该模型能够适应多种类型的材料,并有效预测在不同环境条件(如温度和压力的变化)下的行为。
4、模型稳健性
T-AIMD 在处理带有噪声和不确定性的数据时表现出较高的稳健性。在对比实验中,即使在数据存在轻微偏差的情况下,T-AIMD 也能保持高度的预测精度,这是其他简单机器学习模型难以达到的。
5、扩展性和适应性
T-AIMD 模型的架构允许灵活的调整和优化,适应不断变化的研究需求和新科学发现。这种扩展性使得 T-AIMD 在未来研究中能够持续发挥关键作用,其应用不限于固态电解质,还可拓展到其他能源材料和复杂化学系统的研究。
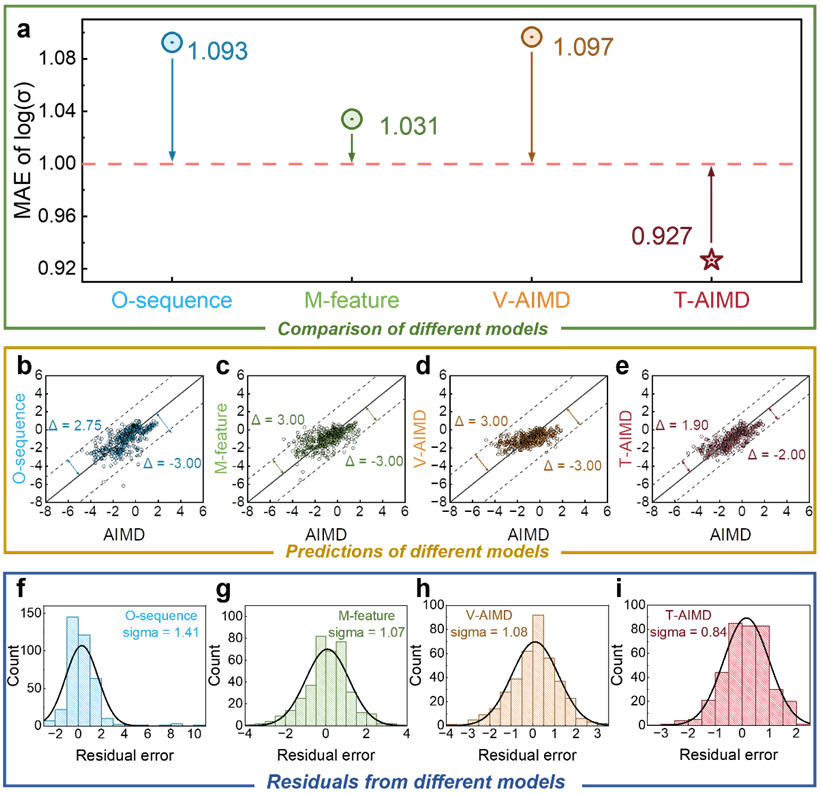
综上所述,基于 T-AIMD 框架,可大幅度加快分子动力学的模拟效率,提升效率 1000 倍、10000 倍、甚至更多,为材料制造和生物设计节省大量的时间成本。
T-AIMD 模型在多个关键方面均优于传统 AIMD 模拟和其他机器学习方法,文本给出的例子显示了其在固态电解质研究和开发中的强大潜力和应用前景。
T-AIMD 的实用性远不止于此。该模型的强大功能和灵活性使其能广泛应用于材料科学的多个领域。
未来,它有望可以用于预测其他类型材料如半导体、金属和高分子材料中的离子和分子行为。
此外,T-AIMD 模型的能力不限于单一离子种类的行为预测,它还能处理多离子系统中的复杂交互作用和动力学问题,这使得它在设计新材料和改善现有材料的性能方面具有极高的实用价值。
以上是计算效率提升100倍以上,上交李金金团队开发基于Transformer的大模型用于从头算分子动力学的详细内容。更多信息请关注PHP中文网其他相关文章!