



编辑 | 枯叶蝶
大型语言模型极大地增强了科学家理解生物学和化学的能力,但基于结构的药物发现、量子化学和结构生物学的可靠方法仍然很少。大型语言模型迫切需要精确的生物分子-配体相互作用数据集。
为了解决这个问题,德国亥姆霍兹慕尼黑研究中心生物学所和慕尼黑工业大学的研究人员,提出了 MISATO。这是一个数据集,它结合了小分子的量子力学(QM)特性,还有约 20,000 个实验蛋白质-配体复合物的相关分子动力学(MD)模拟,以及对实验数据的广泛验证。
从现有的实验结构出发,研究人员利用半经验量子力学系统地完善了这些结构。其中包括大量蛋白质-配体复合物在纯水中的分子动力学模拟,累积时间超过170微秒。
该团队提供了机器学习(ML)基线模型的示例,证明通过使用该数据集可以提高准确性。为机器学习专家提供了一个简单的切入点,以实现下一代药物发现人工智能模型。
该研究以「MISATO: machine learning dataset of protein–ligand complexes for structure-based drug discovery」为题,于 2024 年 5 月 10 日发布在《Nature Computational Science》。

近年来,AI预测技术在科学领域引发了革命,如AlphaFold能精准预测蛋白质结构。尽管结构导向药物发现仍是巨大挑战,AI在此领域的应用尚浅。当前方法面临精度、计算成本及实验依赖度等难题,并多集中于简单解决方案与一维数据处理。忽视了三维蛋白-配体复合体的复杂性。
虽然存在多种数据库,但因数据量限制和热力学信息缺失尚未有AI模型能显示推进药物发现。与AlphaFold在蛋白结构预测领域的成就不同,AI模型还受限于忽视动态性、化学复杂性等问题,影响了其在生物分子分析和量子化学上的潜力。
在这里,德国亥姆霍兹慕尼黑研究中心结构生物学所和慕尼黑工业大学的研究人员,提出了一个基于实验蛋白质-配体结构的蛋白质-配体结构数据库,MISATO(Molecular Interactions Are Structurally Optimized)。
研究人员表明,该数据库有助于更好地训练与药物发现相关领域及其他领域的模型。这包括量子化学、普通结构生物学和生物信息学。

该团队提供了基于量子化学的结构管理和细化,包括配体几何形状的正则化。研究人员用缺失的动态和化学信息来扩充这个数据库,包括时间尺度上的 MD,允许检测某些系统的瞬态和神秘状态。后者对于成功的药物设计非常重要。

因此,研究人员用最大数量的物理参数补充实验数据。这减轻了人工智能模型隐式学习所有这些信息的负担,从而可以专注于主要学习任务。MISATO 数据库提供了一种用户友好的格式,可以直接导入到机器学习代码中。

该团队还提供了各种预处理脚本来过滤和可视化数据集。而且,提供了示例 AI 基线模型,用于计算量子化学性质(化学硬度和电子亲和力)、结合亲和力计算以及预测蛋白质灵活性或诱导拟合特征,从而使数据可以被简化采用。并且,QM、MD 和 AI 模型在实验数据上得到了广泛的验证。
研究人员希望将 MISATO 转变为一个有益的社区项目,造福整个药物发现领域。
论文链接:https://www.nature.com/articles/s43588-024-00627-2
以上是结合量子特征、2万个分子动力学模拟,新蛋白-配体复合物ML数据集,登Nature子刊的详细内容。更多信息请关注PHP中文网其他相关文章!

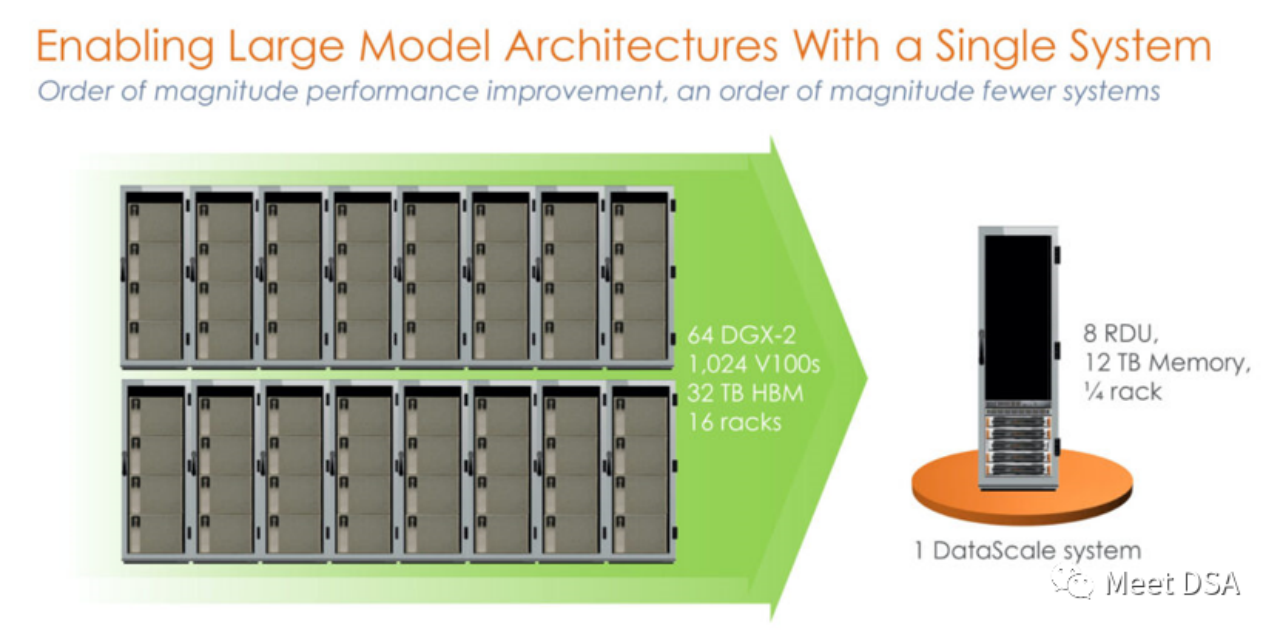 DSA如何弯道超车NVIDIA GPU?Sep 20, 2023 pm 06:09 PM
DSA如何弯道超车NVIDIA GPU?Sep 20, 2023 pm 06:09 PM你可能听过以下犀利的观点:1.跟着NVIDIA的技术路线,可能永远也追不上NVIDIA的脚步。2.DSA或许有机会追赶上NVIDIA,但目前的状况是DSA濒临消亡,看不到任何希望另一方面,我们都知道现在大模型正处于风口位置,业界很多人想做大模型芯片,也有很多人想投大模型芯片。但是,大模型芯片的设计关键在哪,大带宽大内存的重要性好像大家都知道,但做出来的芯片跟NVIDIA相比,又有何不同?带着问题,本文尝试给大家一点启发。纯粹以观点为主的文章往往显得形式主义,我们可以通过一个架构的例子来说明Sam
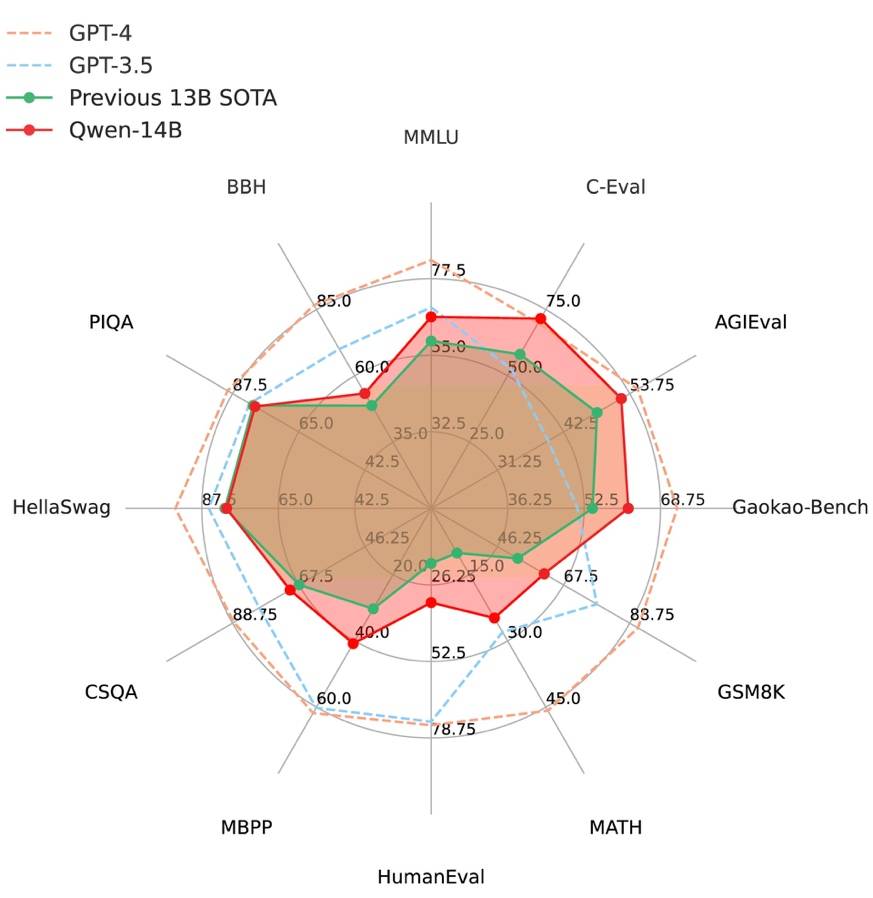 阿里云通义千问14B模型开源!性能超越Llama2等同等尺寸模型Sep 25, 2023 pm 10:25 PM
阿里云通义千问14B模型开源!性能超越Llama2等同等尺寸模型Sep 25, 2023 pm 10:25 PM2021年9月25日,阿里云发布了开源项目通义千问140亿参数模型Qwen-14B以及其对话模型Qwen-14B-Chat,并且可以免费商用。Qwen-14B在多个权威评测中表现出色,超过了同等规模的模型,甚至有些指标接近Llama2-70B。此前,阿里云还开源了70亿参数模型Qwen-7B,仅一个多月的时间下载量就突破了100万,成为开源社区的热门项目Qwen-14B是一款支持多种语言的高性能开源模型,相比同类模型使用了更多的高质量数据,整体训练数据超过3万亿Token,使得模型具备更强大的推
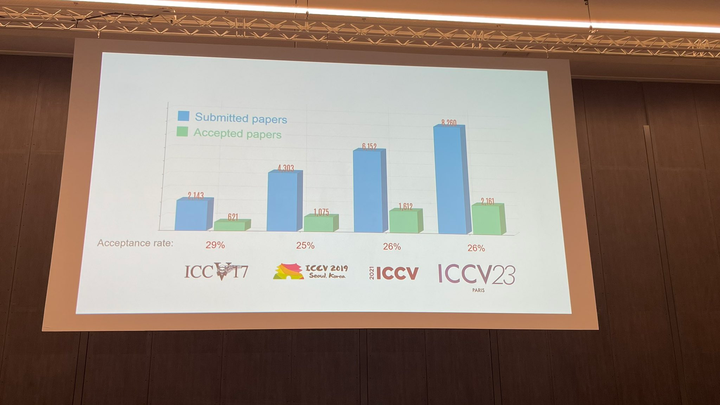 ICCV 2023揭晓:ControlNet、SAM等热门论文斩获奖项Oct 04, 2023 pm 09:37 PM
ICCV 2023揭晓:ControlNet、SAM等热门论文斩获奖项Oct 04, 2023 pm 09:37 PM在法国巴黎举行了国际计算机视觉大会ICCV(InternationalConferenceonComputerVision)本周开幕作为全球计算机视觉领域顶级的学术会议,ICCV每两年召开一次。ICCV的热度一直以来都与CVPR不相上下,屡创新高在今天的开幕式上,ICCV官方公布了今年的论文数据:本届ICCV共有8068篇投稿,其中有2160篇被接收,录用率为26.8%,略高于上一届ICCV2021的录用率25.9%在论文主题方面,官方也公布了相关数据:多视角和传感器的3D技术热度最高在今天的开
 复旦大学团队发布中文智慧法律系统DISC-LawLLM,构建司法评测基准,开源30万微调数据Sep 29, 2023 pm 01:17 PM
复旦大学团队发布中文智慧法律系统DISC-LawLLM,构建司法评测基准,开源30万微调数据Sep 29, 2023 pm 01:17 PM随着智慧司法的兴起,智能化方法驱动的智能法律系统有望惠及不同群体。例如,为法律专业人员减轻文书工作,为普通民众提供法律咨询服务,为法学学生提供学习和考试辅导。由于法律知识的独特性和司法任务的多样性,此前的智慧司法研究方面主要着眼于为特定任务设计自动化算法,难以满足对司法领域提供支撑性服务的需求,离应用落地有不小的距离。而大型语言模型(LLMs)在不同的传统任务上展示出强大的能力,为智能法律系统的进一步发展带来希望。近日,复旦大学数据智能与社会计算实验室(FudanDISC)发布大语言模型驱动的中
 百度文心一言全面向全社会开放,率先迈出重要一步Aug 31, 2023 pm 01:33 PM
百度文心一言全面向全社会开放,率先迈出重要一步Aug 31, 2023 pm 01:33 PM8月31日,文心一言首次向全社会全面开放。用户可以在应用商店下载“文心一言APP”或登录“文心一言官网”(https://yiyan.baidu.com)进行体验据报道,百度计划推出一系列经过全新重构的AI原生应用,以便让用户充分体验生成式AI的理解、生成、逻辑和记忆等四大核心能力今年3月16日,文心一言开启邀测。作为全球大厂中首个发布的生成式AI产品,文心一言的基础模型文心大模型早在2019年就在国内率先发布,近期升级的文心大模型3.5也持续在十余个国内外权威测评中位居第一。李彦宏表示,当文心
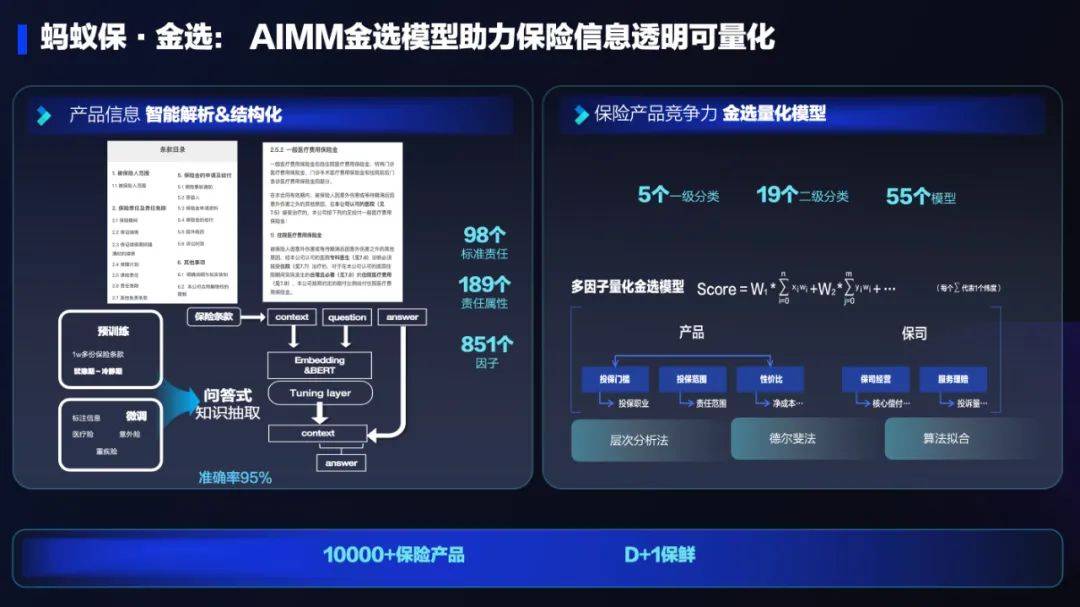 AI技术在蚂蚁集团保险业务中的应用:革新保险服务,带来全新体验Sep 20, 2023 pm 10:45 PM
AI技术在蚂蚁集团保险业务中的应用:革新保险服务,带来全新体验Sep 20, 2023 pm 10:45 PM保险行业对于社会民生和国民经济的重要性不言而喻。作为风险管理工具,保险为人民群众提供保障和福利,推动经济的稳定和可持续发展。在新的时代背景下,保险行业面临着新的机遇和挑战,需要不断创新和转型,以适应社会需求的变化和经济结构的调整近年来,中国的保险科技蓬勃发展。通过创新的商业模式和先进的技术手段,积极推动保险行业实现数字化和智能化转型。保险科技的目标是提升保险服务的便利性、个性化和智能化水平,以前所未有的速度改变传统保险业的面貌。这一发展趋势为保险行业注入了新的活力,使保险产品更贴近人民群众的实际
 致敬TempleOS,有开发者创建了启动Llama 2的操作系统,网友:8G内存老电脑就能跑Oct 07, 2023 pm 10:09 PM
致敬TempleOS,有开发者创建了启动Llama 2的操作系统,网友:8G内存老电脑就能跑Oct 07, 2023 pm 10:09 PM不得不说,Llama2的「二创」项目越来越硬核、有趣了。自Meta发布开源大模型Llama2以来,围绕着该模型的「二创」项目便多了起来。此前7月,特斯拉前AI总监、重回OpenAI的AndrejKarpathy利用周末时间,做了一个关于Llama2的有趣项目llama2.c,让用户在PyTorch中训练一个babyLlama2模型,然后使用近500行纯C、无任何依赖性的文件进行推理。今天,在Karpathyllama2.c项目的基础上,又有开发者创建了一个启动Llama2的演示操作系统,以及一个
 快手黑科技“子弹时间”赋能亚运转播,打造智慧观赛新体验Oct 11, 2023 am 11:21 AM
快手黑科技“子弹时间”赋能亚运转播,打造智慧观赛新体验Oct 11, 2023 am 11:21 AM杭州第19届亚运会不仅是国际顶级体育盛会,更是一场精彩绝伦的中国科技盛宴。本届亚运会中,快手StreamLake与杭州电信深度合作,联合打造智慧观赛新体验,在击剑赛事的转播中,全面应用了快手StreamLake六自由度技术,其中“子弹时间”也是首次应用于击剑项目国际顶级赛事。中国电信杭州分公司智能亚运专班组长芮杰表示,依托快手StreamLake自研的4K3D虚拟运镜视频技术和中国电信5G/全光网,通过赛场内部署的4K专业摄像机阵列实时采集的高清竞赛视频,


热AI工具

Undresser.AI Undress
人工智能驱动的应用程序,用于创建逼真的裸体照片

AI Clothes Remover
用于从照片中去除衣服的在线人工智能工具。

Undress AI Tool
免费脱衣服图片

Clothoff.io
AI脱衣机

AI Hentai Generator
免费生成ai无尽的。

热门文章
热工具

Dreamweaver Mac版
视觉化网页开发工具

DVWA
Damn Vulnerable Web App (DVWA) 是一个PHP/MySQL的Web应用程序,非常容易受到攻击。它的主要目标是成为安全专业人员在合法环境中测试自己的技能和工具的辅助工具,帮助Web开发人员更好地理解保护Web应用程序的过程,并帮助教师/学生在课堂环境中教授/学习Web应用程序安全。DVWA的目标是通过简单直接的界面练习一些最常见的Web漏洞,难度各不相同。请注意,该软件中

Dreamweaver CS6
视觉化网页开发工具

EditPlus 中文破解版
体积小,语法高亮,不支持代码提示功能

SublimeText3 Linux新版
SublimeText3 Linux最新版

