



windows系統能不能裝hmmer軟體
hmmer下載與安裝
對於Mac OS/X, Linux, UNIX系統,用原始碼編譯安裝:
% wget ftp://selab.janelia.org/pub/software/hmmer3/3.0/hmmer-3.0.tar.gz % tar zxf hmmer-3.0.tar.gz % cd hmmer-3.0 % ./configure % make % make check
windows系統,直接下載二進位壓縮包,解壓縮就可以使用。
hmmer包含的程式
phmmer: 與Blastp類似,使用一個蛋白質序列搜尋蛋白質序列庫;
>phmmer tutorial/HBB HUMAN uniprot sprot.fa
jackhmmer: 與psiBlast類似,蛋白質序列迭代搜尋蛋白質序列庫;
>jackhmmer tutorial/HBB HUMAN uniprot sprot.fa
hmmbuild: 用多重比對序列建構HMM模型;
hmmsearch: 使用HMM模型搜尋序列庫;
hmmscan: 使用序列搜尋HMM函式庫;
hmmalign: 使用HMM為線索,建構多重比對序列;
>hmmalign globins4.hmm tutorial/globins45.fa
hmmconvert: 轉換HMM格式
hmmemit: 從HMM模型中,得到一個模式序列;
hmmfetch: 透過名字或接受號從HMM庫取回一個HMM模型;
hmmpress:格式化HMM資料庫,以便於hmmscan搜尋使用;
hmmstat:顯示HMM資料庫的統計資料;
使用HMM模型搜尋序列資料庫
使用hmmbuild建構HMM模型,輸入為Stockholm格式或FASTA格式的多重比對序列檔(如:tutorial/globins4.sto),指令如下:
>hmmbuild globins4.hmm tutorial/globins4.sto
globins4.hmm為輸出的HMM模型
使用hmmsearch搜尋蛋白質序列資料庫,蛋白質序列資料庫為FASTA格式,指令如下:
>hmmsearch globins4.hmm uniprot sprot.fasta >globins4.out
globins4.out為輸出的結果文件,如下:
*範例使用官方教學中的範例
使用蛋白質序列搜尋HMM資料庫
建立HMM資料庫,HMM資料庫是包含多個HMM模型的文件,可以從Pfam、SMART、TIGRFams下載,也可以自己由多重比對序列集中構建,如:
>hmmbuild globins4.hmm tutorial/globins4.sto
>hmmbuild fn3.hmm tutorial/fn3.sto
>hmmbuild Pkinase.hmm tutorial/Pkinase.sto
>cat globins4.hmm fn3.hmm Pkinase.hmm >minifam
#使用hmmpress格式化資料庫,包括壓縮以及建立索引,指令如下:
>hmmpress minifam
這個步驟可以很快的執行完成,輸出的內容如下:
Working… done.
Pressed and indexed 3 HMMs (3 names and 2 accessions).
Models pressed into binary file: minifam.h3m
SSI index for binary model file: minifam.h3i
Profiles (MSV part) pressed into: minifam.h3f
Profiles (remainder) pressed into: minifam.h3p
使用hmmscan搜尋HMM資料庫,指令如下:
>hmmscan minifam tutorial/7LESS_DROME
hmmer軟體怎麼將fasta格式檔案轉換為sto格式
這問題我也遇到了,網路找半天沒找到合適的方案,於是自己寫了一個,程式碼如下
import glob # 都是標準函式庫的東西
import os
# 把你想建造hmm的fasta檔案(比對好的)和本程式放在同一個資料夾裡,然後執行本程式直接跑hmmbuild
os.chdir(os.path.dirname(__file__))
fs = glob.glob('*.fasta') # 取得每個fasta文件,如果你的fasta檔案裡有不是.fasta後綴名的,可以改這裡,或直接改成'*.fa*'
for f in fs:
hmm = os.path.splitext(f)[0] '.hmm'
stockholm = os.path.splitext(f)[0] '.sto'
with open(f, 'r') as fhandle: # 這個是讀fasta檔案用的,把所有fasta檔案都存到列表裡
fastas = ['>' tmp.replace('\n', '\r', 1).replace('\n', '').replace('\r', '\n') for tmp in tuple(filter(None, (fhandle.read().split('>'))))]
for i in range(len(fastas)):
fastas[i] = fastas[i].split('\n')
fastas[i][0] = fastas[i][0].split()[0][1:10]
tmp = []
for j in range(len(fastas[i][1]) // 80 1):
tmp.append(fastas[i][1][80 * j : 80 * j 80])
fastas[i][1] = tmp
with open(stockholm, 'w') as out: # 這裡在寫sto檔案
out.write('# STOCKHOLM 1.0\n\n')
for j in range(len(fastas[0][1]) - 1):
for i in range(len(fastas)):
out.write('% -12s%s\n' % (fastas[i][0], fastas[i][1][j]))
out.write('\n')
for i in range(len(fastas)):
out.write('% -12s%s\n' % (fastas[i][0], fastas[i][1][-1]))
out.write('//')
os.system('hmmbuild --amino %s %s' % (hmm, stockholm)) # 這裡在跑hmmbuild,你可以自行修改裡面的參數
如何自學生物資訊學
1,從現有的生物資訊工具開始,要熟悉如何利用先用的軟體、網路伺服器、資料庫等等,為生物研究服務,不要做重複工作,能用現成的就不自己開發。
2,熟悉命令列的作業系統,DOS,Linux,可以寫簡單的shell;進而安裝命令列層級的程序,跑一些常規的流程。要學習如何尋找和安裝軟體,這是最重要且最基本的技能。其實很多問題,如果找到合適的軟體包,都是迎刃而解的。
3,熟悉一種簡單的腳本語言,個人推薦用python,具體原因可以見我的貼文。在沒有現成工具時,或需要資料格式轉換時,小的腳本是非常有用的。一般的應用程式不需要自己寫太多的程式碼,要相信我們通常遇到的問題,別的高手可能早就遇到了,所以網路上有大量的工具包。至於更多的程式語言,一門精則門門通,R,perl等都是類似的。
4,熟悉簡單的演算法和資料結構的知識,這樣就可以理解許多程式的內在機制,進而知道它們的優點和缺點,對自己寫程式也有幫助。有精力的話,進而學習統計、機器學習等。 。
5,在自己的生物領域內擴展,調查,分析,開發。
以上是能否在Windows系統上安裝HMMER軟體?的詳細內容。更多資訊請關注PHP中文網其他相關文章!

 我想念時代的計算機是插頭和播放May 13, 2025 am 06:03 AM
我想念時代的計算機是插頭和播放May 13, 2025 am 06:03 AM建造一台新電腦曾經是勝利的時刻。現在,這僅僅是開始,因為您必須安裝應用程序,下載大型遊戲並在完全設置之前調整數十個設置。讓我們看一下我們如何到達這里以及過去的情況。
 為什麼這款Logitech鼠標在每個技術人員的桌子上?May 13, 2025 am 06:02 AM
為什麼這款Logitech鼠標在每個技術人員的桌子上?May 13, 2025 am 06:02 AMLogitech的MX Master系列小鼠已經成為YouTube視頻或精通技術人員的桌子設置的照片,但是是什麼使現在的Iconic鼠標如此特別? 目前,我在我的第三個MX Master Mouse上,老實說,我可以
 我拒絕在沒有屏幕的情況下購買鍵盤May 13, 2025 am 03:05 AM
我拒絕在沒有屏幕的情況下購買鍵盤May 13, 2025 am 03:05 AM機械鍵盤提供了很多功能,從開關類型和鍵蓋材料到聲音衰減。 但是,內置的屏幕經常被忽略但寶貴的功能。 為什麼將屏幕集成到鍵盤中? QWERTY鍵盤,主食
 Galaxy S25變得前衛和RIP Skype:每週綜述May 13, 2025 am 03:04 AM
Galaxy S25變得前衛和RIP Skype:每週綜述May 13, 2025 am 03:04 AM技術新聞綜述:整個技術領域的重大更新和新版本 本週,從AI的進步到新遊戲外圍設備和重大軟件更新。讓我們深入亮點: AI和SECU
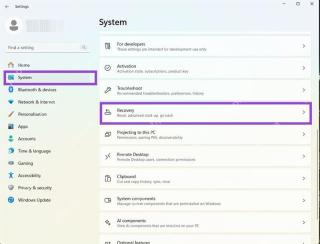 如何回滾Windows 11更新May 12, 2025 pm 08:01 PM
如何回滾Windows 11更新May 12, 2025 pm 08:01 PMWindows 11 更新導致系統問題?別慌!本文提供三種回滾更新的方法,助您恢復系統穩定性。 方法一:通過 Windows 設置回滾更新 此方法適用於更新時間在 10 天以內的用戶。 步驟 1: 點擊“開始”菜單,進入“設置”。您也可以按鍵盤上的 Windows 鍵 I。 步驟 2: 在“設置”中,選擇“系統”,然後點擊“恢復”。 步驟 3: 在“恢復選項”下,找到“以前的 Windows 版本”。如果“返回”按鈕可點擊,則可將系統回滾到之前的版本。 步驟 4: 系統會詢問您回滾的原因
 13個Windows鍵盤快捷鍵May 12, 2025 am 03:02 AM
13個Windows鍵盤快捷鍵May 12, 2025 am 03:02 AM掌握Windows鍵盤快捷鍵不僅僅是效率;它簡化了您的整個計算體驗。 Windows的界面可能不如直觀,隱藏在菜單層中的關鍵設置。 幸運的是,存在無數捷徑
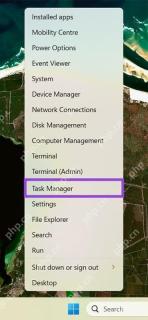 如何加快PC(Windows 11)May 11, 2025 pm 06:01 PM
如何加快PC(Windows 11)May 11, 2025 pm 06:01 PM您的Windows 11 PC的運行速度比平常慢嗎? 打開應用程序和加載網站佔據永恆?你並不孤單! 本指南提供了三個簡單的無下載解決方案,可以在沒有復雜設置調整的情況下提高計算機的性能
 這款迷你PC兼作不太好的平板電腦May 11, 2025 am 06:01 AM
這款迷你PC兼作不太好的平板電腦May 11, 2025 am 06:01 AM這台迷你PC偽裝成平板電腦,還有很多不足之處。 7英寸,1290x800分辨率的屏幕令人難以置信。雖然有些人可能將其用於媒體消費(類似於7英寸的亞馬遜消防片),但不太可能是主要選擇


熱AI工具

Undresser.AI Undress
人工智慧驅動的應用程序,用於創建逼真的裸體照片

AI Clothes Remover
用於從照片中去除衣服的線上人工智慧工具。

Undress AI Tool
免費脫衣圖片

Clothoff.io
AI脫衣器

Video Face Swap
使用我們完全免費的人工智慧換臉工具,輕鬆在任何影片中換臉!

熱門文章
熱工具

SublimeText3漢化版
中文版,非常好用

MinGW - Minimalist GNU for Windows
這個專案正在遷移到osdn.net/projects/mingw的過程中,你可以繼續在那裡關注我們。 MinGW:GNU編譯器集合(GCC)的本機Windows移植版本,可自由分發的導入函式庫和用於建置本機Windows應用程式的頭檔;包括對MSVC執行時間的擴展,以支援C99功能。 MinGW的所有軟體都可以在64位元Windows平台上運作。

SublimeText3 Mac版
神級程式碼編輯軟體(SublimeText3)

Dreamweaver CS6
視覺化網頁開發工具

Atom編輯器mac版下載
最受歡迎的的開源編輯器

