
威大華人團隊全新多模態資料分析及生成方法JAMIE,大幅提升細胞類型、功能預測能力

- WBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWB轉載
- 2023-06-10 14:43:291454瀏覽
近年來,隨著單細胞技術的迅速發展,我們得以測量了單一細胞的各種特徵從而獲取單細胞多模態資料(例如scRNA-seq,scATAC-seq,Patch-seq) 。
這些數據有助於我們深入了解細胞功能和分子機制。例如研究人員近來多透過機器學習方法分析單細胞多模態資料間的關係,進而理解細胞類型和疾病所涉及的生物機制。
但是單細胞多模態資料的取得常常成本高昂,且模態缺失時有發生。而現有的機器學習方法通常需要完全匹配的多模態資料才能進行資料填補和嵌入,不適用於模態缺失的情況。
為了解決這個問題,美國威斯康辛大學麥迪遜分校王岱峰實驗室開發了一種基於聯合變分自動編碼器的開源機器學習方法——Joint Variational Autoencoders for Multimodal Imputation and Embedding(JAMIE)。
JAMIE可用於單細胞多模態資料整合分析,如資料對齊、嵌入,和對遺失資料進行添補,以便更好的預測細胞類型及功能。
此工作已於近日發表於《自然–機器智能》(Nature Machine Intelligence)。

論文網址:https://www.nature.com/articles/s42256-023-00663 -z
專案網址:https://github.com/daifengwanglab/JAMIE
##JAMIE方法介紹JAMIE訓練了一種可重複使用的聯合變分自編碼器模型,將可用的多模態資料分別投影到相似的潛空間中,從而增強了單模態模式的推斷能力。
如圖1所示,為了執行跨模態填補,JAMIE將資料饋入編碼器,然後將潛空間結果透過相反的解碼器進行處理。
JAMIE將自編碼器的可重複使用和靈活的潛空間產生與對齊方法的自動對應估計相結合,從而能夠處理不完全對應的多模態資料。
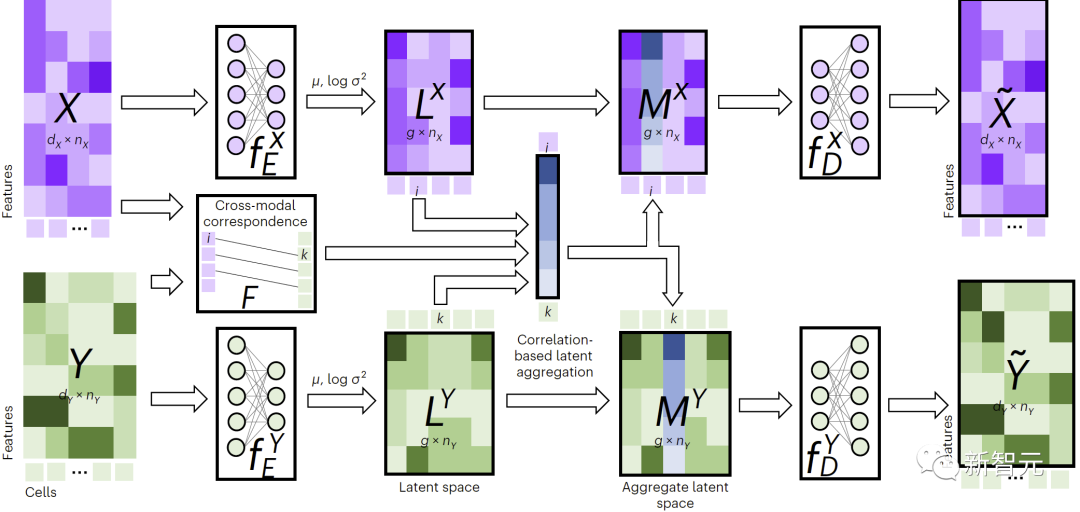
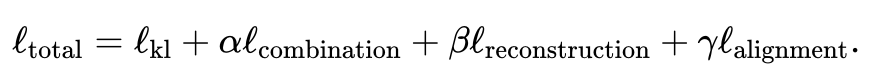
###################### #具體而言,JAMIE可以分為以下兩步驟:############輸入資料預處理。以雙模態為例,假設模態對應資料矩陣分別為和。注意這裡特徵維度和可以不同,樣本數目和也可以不同。預處理對每個矩陣的每一行都歸一化成均值0和方差1。如果有對應數據,使用者可以提供模態相關矩陣來改善效能,其中 表示模態中的第個樣本和模態中的第個樣本完全對應,表示沒有已知的對應關係,表示有部分的對應關係。 ######利用聯合變分自編碼器學習每個模態的相似潛空間: 和 ,其中(默認,用戶可調節)是潛空間維度。訓練過程中,JAMIE最小化如下損失函數:############################總損失函數包含四項。 ############其中第一項計算變分自編碼器推斷出的分佈與多元標準常態分佈之間的Kullback-Leibler (KL)散度,有助於維持潛空間的連續性;第二項強制對應樣本的相似性;第三項是重構資料矩陣和原始資料矩陣之間的平均平方誤差和;第四項利用推論的跨模態對應關係來調整產生的潛空間。 ######
各項的具體表達方式見論文原文。第二、三、四項的相對第一項的權重可由使用者自行調節,JAMIE也提供了可適用於常用情況的預設權重。
下述表格展示了JAMIE與目前最先進方法的模型和適用範圍的比較。 JAMIE將幾種不同的整合和插補方法的特徵統一到一個單一的架構中,因此能夠進行缺失模態插值,從而具有非組學數據兼容性、且能處理只有部分對應關係的多模態數據的優點。
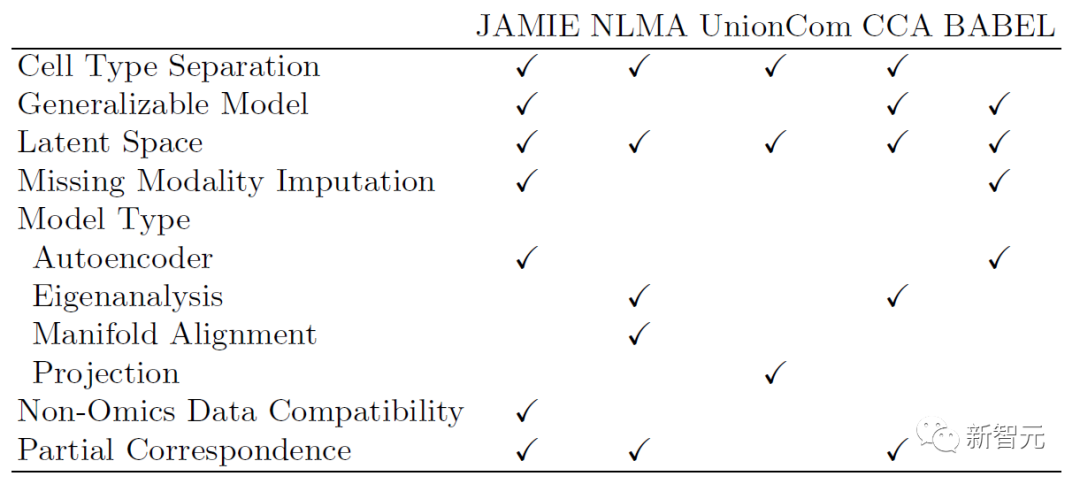
#表1. 各種多模態整合與缺失模態填補方法的比較。透過單一架構,JAMIE整合了來自多種不同整合和插值方法的特徵。 NLMA:非線性流形對齊[15],UnionCom[7],CCA:典型相關分析[15, 16],BABEL[5]。
JAMIE的主要應用
多模態資料的整合和表型預測
對多模態資料的整合可以改善分類性能、增進對錶型知識和複雜生物機制的理解。
給定兩個資料集、和對應關係,JAMIE可以根據訓練好的編碼器和產生潛空間資料、,並基於、進行聚類或分類。
基於潛空間資料的聚類具有幾個優勢,如將兩種模態都納入特徵生成。然後,JAMIE可以預測樣本對應關係,並如細胞類型預測。
對於部分標註的資料集,同一聚類的細胞們應該具有相似的類型。
JAMIE在產生潛空間資料的過程中就進行了分離了不同類型資料的特徵,因此通常不需要複雜的聚類或分類演算法就可以達到較好的效果。
對於高維度數據,JAMIE使用UMAP[32]進行細胞類型聚類視覺化。
跨模態資料填補
#目前跨模態填補的許多方法不能展示它們學習到了用於填補目的的潛在生物機制。
對比於前饋網路或線性迴歸方法,JAMIE能基於更嚴格的數學基礎更好的學習到潛在的生物機制來預測缺失資料。
圖2展示了JAMIE用於跨模態資料填補的流程。 JAMIE先是針對訓練資料訓練編碼和解碼模型。
對於新資料 ,JAMIE首先利用資料學習到的編碼器將其投影到潛空間得到 ,然後透過聚合潛空間特徵的方法得到 ,最後透過對應的解碼器將解碼成缺失模式的資料。
JAMIE使用潛空間預測細胞的對應關係,這可能有助於理解資料特徵和表型之間的關係。
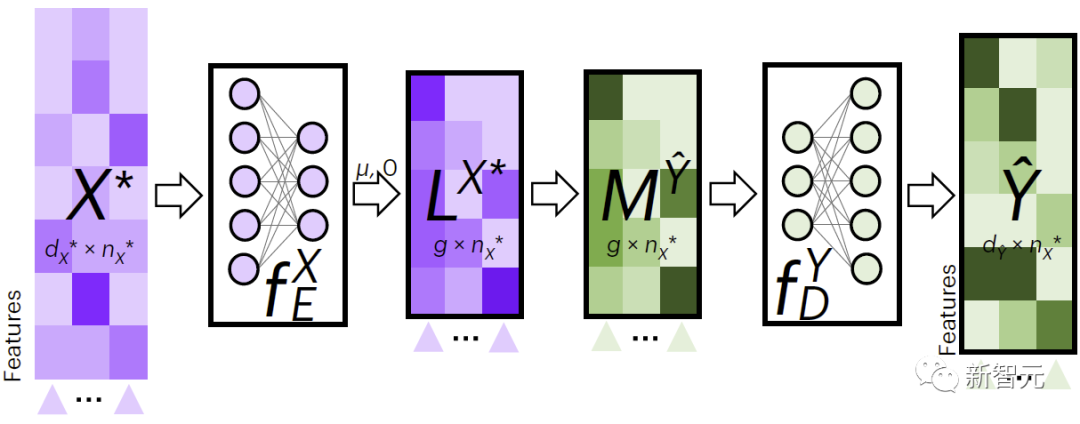
#圖2.JAMIE跨模態插補
潛空間特徵和填補特徵的解釋
為了解釋訓練的模型,JAMIE採用了SHAP(SHapley Additive exPlanations )[18]。
SHAP透過對模型產生的個體預測進行樣本調變來評估各個輸入特徵的重要性。這可以用於各種有趣的應用。
如果目標變數可以透過表型輕鬆分離,SHAP可以確定進一步研究的相關特徵。此外,如果我們進行填補,SHAP可以揭示模型學到的跨模態聯繫。
給定模型和樣本,學習到SHAP值,使得,其中是背景特徵向量。
如果,則SHAP值的總和和背景輸出將等於,其中每個與對模型輸出的影響成比例。
另一種有用的技術是選擇一個關鍵指標用於分類(例如,LTA[7,19])或填補(例如,填補特徵與測量特徵之間的對應關係),並在模型中逐一移除(以背景值取代)每個特徵來評估該指標。
然後,如果關鍵指標變得更糟,這表示被移除的特徵對於模型的結果更為重要。
實驗結果
JAMIE採用了四個常用的單細胞多模態資料集進行驗證。
(1)來自MMD-MA的分支流形的高斯分佈採樣產生的模擬多模態資料(300個樣本,3個細胞類型);
(2)來自小鼠視覺皮質(3,654個樣本,6個細胞類型)和小鼠運動皮質(1,208個樣本,9個細胞類型)的單一神經元細胞的Patch-seq基因表現和電生理特徵特徵資料;
(3)來自人類發育中的大腦(21個孕週,覆蓋人類大腦皮質的7種主要細胞類型)中8,981個樣本的10x單細胞多組學基因表現和染色質可及性數據;
(4)來自COLO-320DM結腸腺癌細胞系的4,301個細胞的scRNA-seq基因表達和scATAC-seq染色質可及性資料。
評估發現,JAMIE明顯優於其他方法(如圖三的MMD-MA的分支流形模擬數據結果比較,和圖四小鼠視覺皮層數據結果比較)並優先考慮了多模態填補的重要特徵,同時在細胞分辨率層面上提供了潛在的新機制洞見。
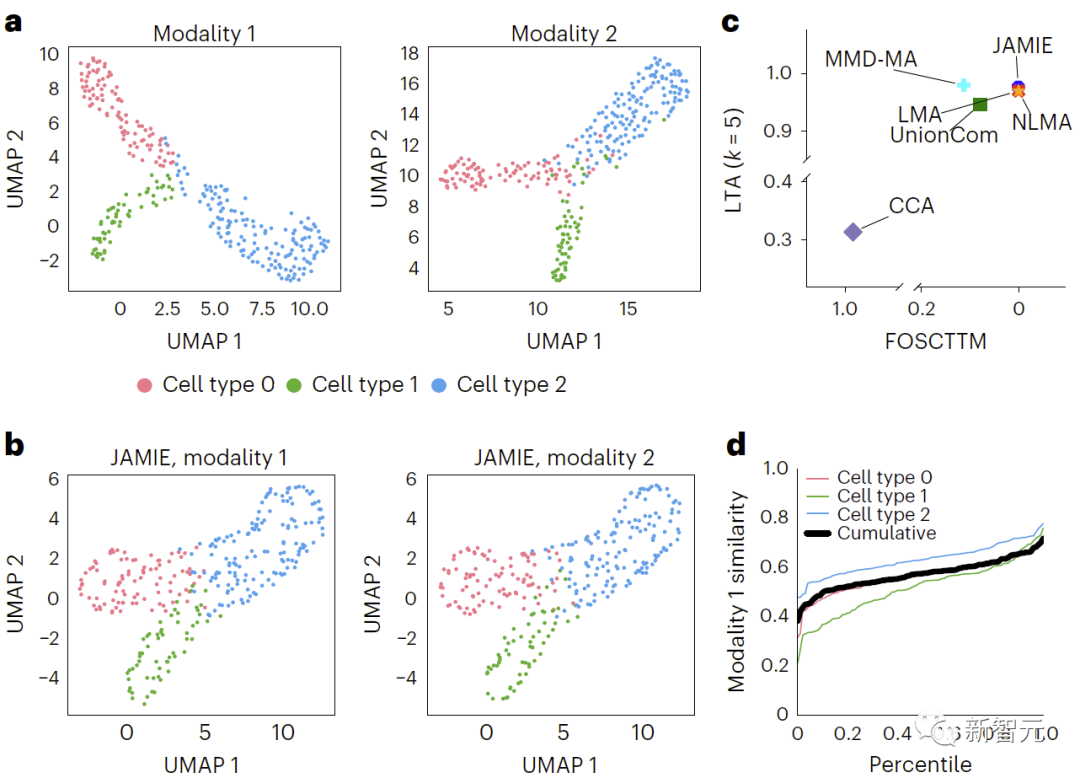
在圖3中,透過對原始空間資料應用UMAP演算法並根據不同細胞類型上色,比較了模擬的多模態資料結果。 b、JAMIE潛在空間的UMAP。 c、JAMIE和現有技術(CCA[15,16],LMA[15],MMD-MA[8],NLMA[15]和UnionCom[7])在使用所有可用的對應資訊進行細胞類型分離時的比較。 x軸為更接近真實平均數的樣本比例,y軸為LTA[7,19]值。在模態1中,計算1-JS距離的累積分佈,評估測量值與插補值的相似性。每條彩線都代表一個特定細胞類型的相似性,而黑線則表示不同細胞類型的平均相似性。
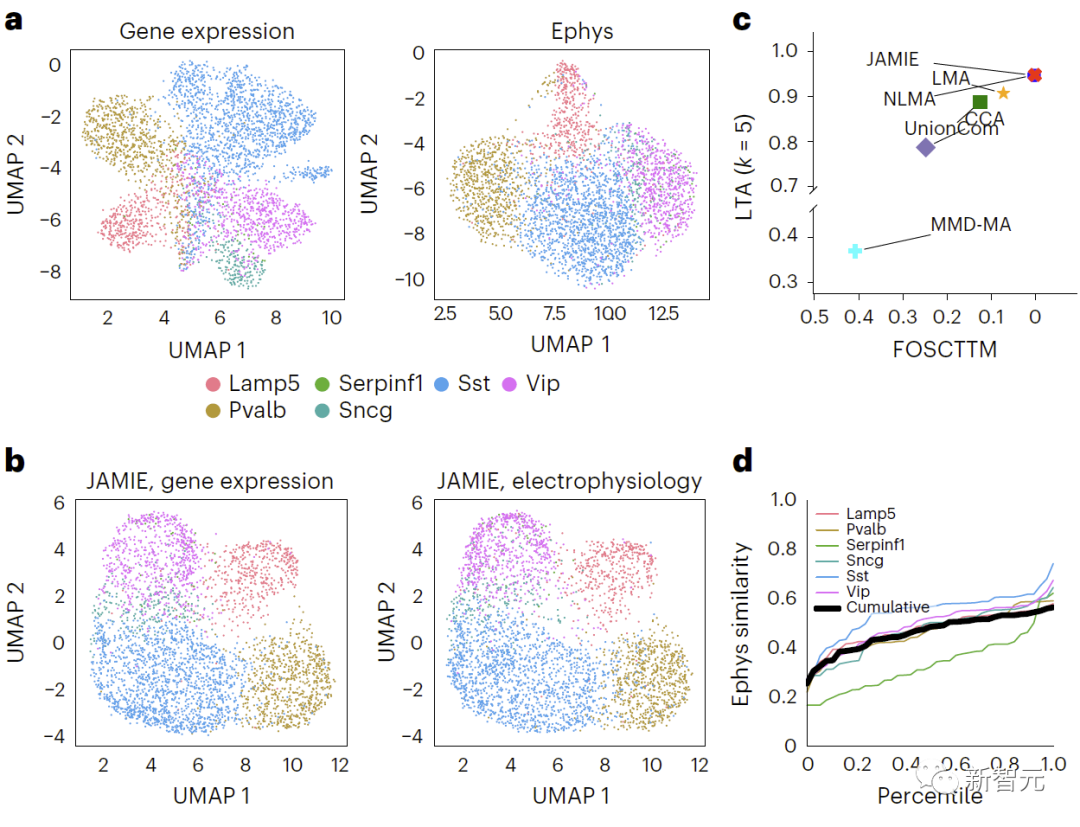
# 重新表達:比較小鼠視皮質中基因表現與電生理特徵結果,使用原始空間中的UMAP,對不同的細胞類型進行著色。圖4展示了該比較結果。 b、JAMIE潛在空間的UMAP。 c、JAMIE和現有技術(CCA[15,16],LMA[15],MMD-MA[8],NLMA[15]和UnionCom[7])在使用所有可用的對應資訊進行細胞類型分離時的比較。 x軸為更接近真實平均數的樣本比例,y軸為LTA[7,19]值。在模態1中,對於1-JS距離計算得出的測量值與插補值之間相似性的累積分佈進行研究。每條彩線代表一種細胞類型的相似性,而黑線表示不同細胞類型的平均相似性。
總結
總而言之,JAMIE 是一種用於單細胞多模態資料整合預測的新型深度神經網路模型。
它適用於複雜、混合或部分對應的多模態數據,透過依賴聯合變分自編碼器(VAE)結構的新穎潛在嵌入聚合方法來實現。除了上述的優越效能外,JAMIE 還具有高效的運算能力和較低的記憶體使用需求。此外,預訓練模型以及學習到的跨模態潛在嵌入可以在下游分析中重複使用。
當然對於較大的資料集,訓練變分自編碼器(VAEs)需要耗費大量時間。因此,JAMIE 中的自動 PCA 等先前特徵選擇方法有助於減輕時間要求。由於VAE使用重建損失,資料預處理也至關重要,以避免大量或重複的特徵對低維嵌入特徵產生不成比例的影響。對於特定的跨模態插補,必須仔細考慮訓練資料集的多樣性,以避免對最終模型產生偏差並對其泛化能力產生負面影響。 JAMIE 還可以潛在地擴展到對來自不同來源而不是不同模態的數據集進行對齊,例如在不同條件下的基因表現數據。
作者介紹
論文作者Noah Cohen Kalafut(電腦系博士生),黃翔(資深研究員),王岱峰(PI)隸屬於威斯康辛大學麥迪遜分校生物統計和醫學資訊學系、電腦科學系和威斯曼研究中心。通訊作者為王岱峰教授。
成立於1973年的威斯曼中心半世紀以來致力於推進人類發育,神經發育障礙和神經退化性疾病的研究。
以上是威大華人團隊全新多模態資料分析及生成方法JAMIE,大幅提升細胞類型、功能預測能力的詳細內容。更多資訊請關注PHP中文網其他相關文章!