
數百萬晶體資料訓練,解決晶體學相位問題,深度學習方法PhAI登Science

- 王林原創
- 2024-08-08 21:22:30950瀏覽

時至今日,晶體學所測定的結構細節和精度,從簡單的金屬到大型膜蛋白,是任何其他方法都無法比擬的。然而,最大的挑戰——所謂的相位問題,仍然是從實驗確定的振幅中檢索相位資訊。
丹麥哥本哈根大學研究人員,開發了一種解決晶體相問題的深度學習方法PhAI,利用數百萬人工晶體結構及其相應的合成衍射數據訓練的深度學習神經網絡,可以產生準確的電子密度圖。
研究表明,這種基於深度學習的從頭算結構解決方案方法,可以以僅2 埃的分辨率解決相位問題,該分辨率僅相當於原子分辨率可用數據的10% 到20%,而傳統的從頭算方法通常需要原子分辨率。
相關研究以《PhAI: A deep-learning approach to solve the crystallographic phase problem》為題,於 8 月 1 日發佈在《Science》上。
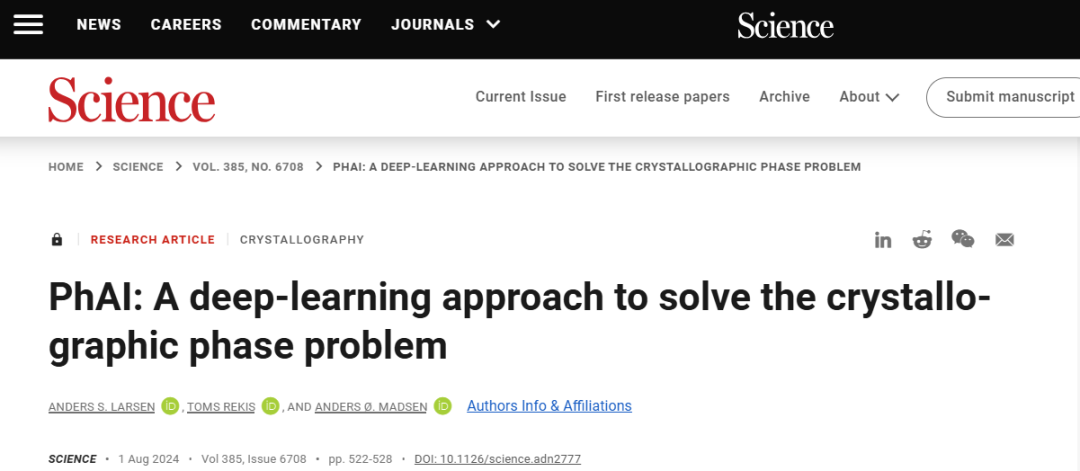
晶體學是自然科學中的核心分析技術之一。 X 射線晶體學為晶體的三維結構提供了獨特的視角。
為了重建電子密度圖,必須知道足夠的衍射反射的複雜結構因子 $F$。在傳統實驗中,只能得到振幅 $|F|$,相位 $phi$ 會遺失。這是晶體學相位問題。
20 世紀 50 年代和 60 年代取得了重大突破,Karle 和 Hauptmann** 開發了用於解決相位問題的所謂直接方法。但直接法需要原子分辨率的繞射數據。然而,原子分辨率的要求是一種經驗觀察。
近年來,傳統的直接方法已被對偶空間方法所補充。 目前可用的從頭算方法似乎已達到極限。相位問題的普遍解決方案仍然未知。
從數學上講,結構因子振幅與相位的任何組合都可以進行逆傅立葉變換。 然而,物理和化學要求(例如具有原子狀電子密度分佈)對與一組振幅一致的相位的可能組合施加了規則。深度學習的進步使得人們能夠探索這種關係,也許比目前的從頭算方法更深入。
在此,哥本哈根大學的研究人員採用了數據驅動的方法,使用數百萬個人造晶體結構及其相應的衍射數據,旨在解決晶體學中的相位問題。
研究表明,這種基於深度學習的從頭算結構解決方案方法,可以在僅最小晶格平面距離(dmin)= 2.0 Å 的分辨率下執行,只需要使用直接方法所需數據的10% 到20%。
神經網路的設計與訓練
所建構的人工神經網路稱為 PhAI,接受結構因子振幅 |F| 並輸出對應的相位值 ϕ ϕ 。 PhAI
的架構如下圖所示。 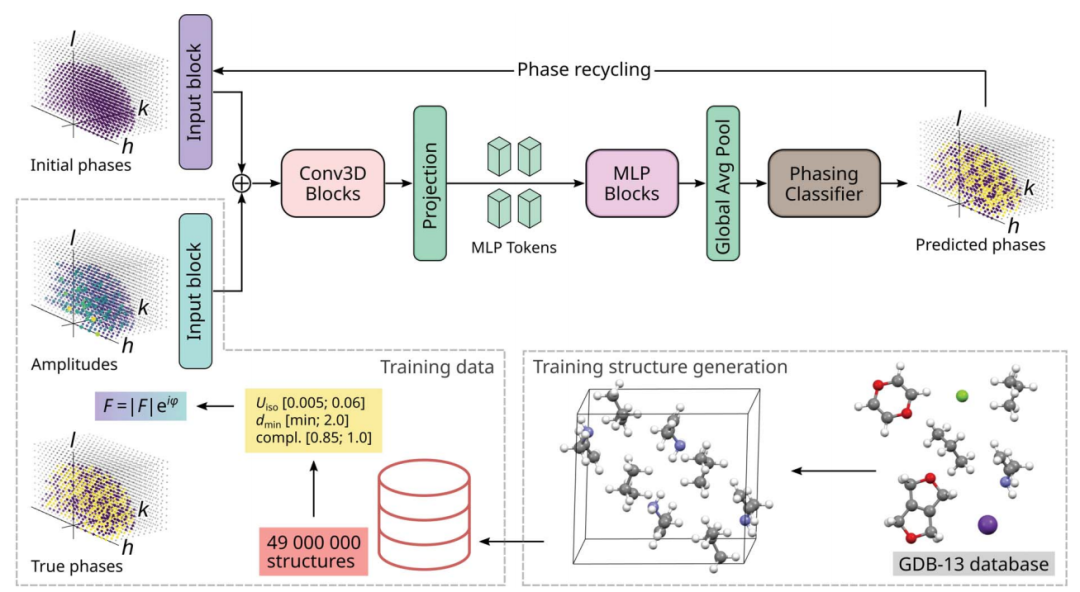
圖示:PhAI 神經網路方法解決相位問題。 (資料來源:論文)晶體結構的結構因子數量取決於晶胞大小。根據計算資源,對輸入資料的大小設定了限制。輸入結構因子振幅是根據 Miller 指數 (h、k、l) 服從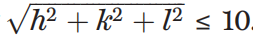
1. 反射來選擇的。 也就是,限制在原子解析度下晶胞尺寸約為 10 Å 的結構。此外,選擇了最常見的中心對稱空間群 P21/c。中心對稱性將可能的相位值限制為零或 π rad。 🎜
- Research on training neural networks using artificial crystal structures containing mainly organic molecules. Approximately 49,000,000 structures were created, of which 94.29% were organic crystal structures, 5.66% were metal-organic crystal structures, and 0.05% were inorganic crystal structures.
- The input to the neural network consists of amplitude and phase, which are processed by a convolutional input block, added and fed into a series of convolutional blocks (Conv3D), followed by a series of multilayer perceptron (MLP) blocks. The predicted phase from the linear classifier (phase classifier) is cycled through the network Nc times. Training data were generated by inserting metal atoms and organic molecules from the GDB-13 database into unit cells. The resulting structures are organized into training data from which the true phase and structure factor amplitudes at sampled temperature factors, resolution and integrity can be calculated.
Solve real structural problems - Trained neural networks run on standard computers with moderate computational requirements. It accepts as input a list of hkl indices and corresponding structure factor amplitudes. No other input information is required, not even the unit cell parameters of the structure. This is fundamentally different from all other modern ab initio methods. The network can predict and output phase values on the fly.
- The researchers tested the performance of the neural network using calculated diffraction data from real crystal structures. A total of 2387 test cases were obtained. For all collected structures, multiple data resolution values ranging from 1.0 to 2.0 Å were considered. For comparison, a charge flip method was also used to retrieve phase information.
Illustration: Histogram of the correlation coefficient r between phase and true electron density map.
(Source: paper)
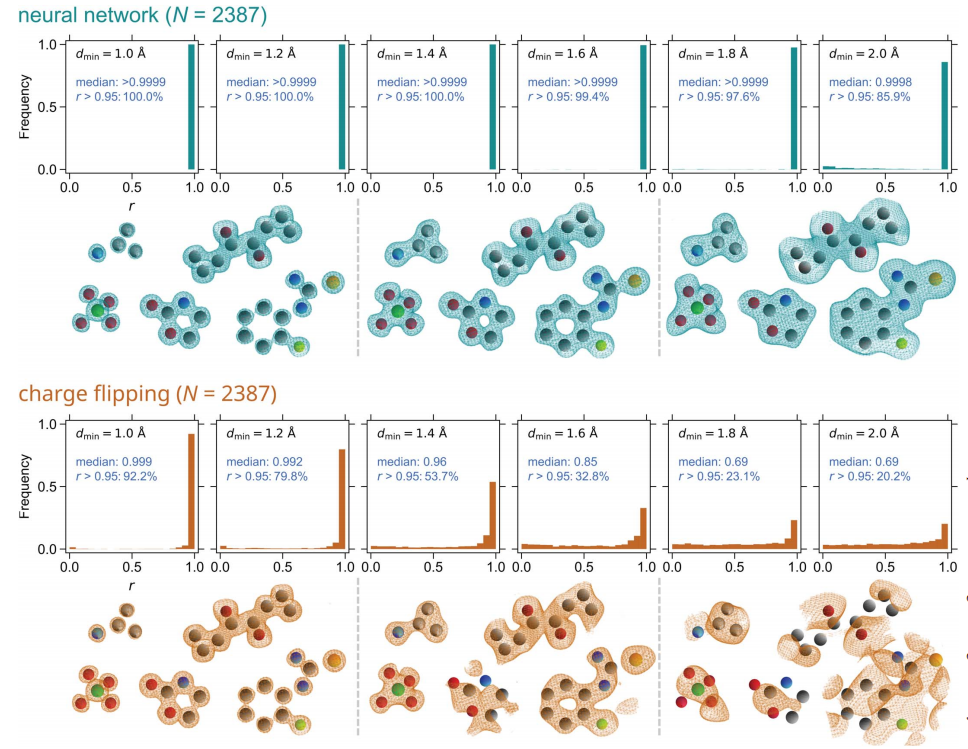
The trained neural network performs well; It can solve all tested structures (N = 2387) if the corresponding diffraction data are of good resolution, and it is better at solving structures from low-resolution data Excellent performance. Although a neural network is rarely trained on inorganic structures, it can solve such structures perfectly.
The charge flip method performs well when processing high-resolution data, but its ability to produce reasonably correct solutions gradually decreases as the data resolution decreases; however, it still solves approximately 32 pixels at a resolution of 1.6Å % Structure. The number of structures identified by charge flipping can be improved by further experimentation and changing input parameters such as flipping thresholds.
In the PhAI approach, This meta-optimization is performed during training and does not need to be performed by the user. These results suggest that the common notion in crystallography that atomic resolution data are necessary to calculate phases ab initio may be broken. PhAI requires only 10% to 20% atomic resolution data.
This result clearly shows that atomic resolution is not necessary for ab initio methods and opens new avenues for deep learning-based structure determination.
The challenge of this deep learning approach is to scale the neural network, that is, diffraction data for larger unit cells will require a large amount of input and output data as well as computational cost during training. In the future, further research is needed to extend this method to the general case.
以上是數百萬晶體資料訓練,解決晶體學相位問題,深度學習方法PhAI登Science的詳細內容。更多資訊請關注PHP中文網其他相關文章!