



編輯 | 蘿蔔皮
蛋白質是身體對抗病原和開發中一種成熟的工具,並用於縮小實驗測試的潛在治療範圍。高品質的蛋白質結構是必需的,並且蛋白質經常被視為完全或部分剛性的。
在這裡,柏林自由大學(Freie Universität Berlin)的研究人員開發了一個人工智慧系統,可以直接從序列資訊預測蛋白質-配體複合物的完全柔性全原子結構。
雖然經典對接方法仍然更勝一籌,但這也取決於目標蛋白質的晶體結構。除了預測靈活的全原子結構外,預測置信度指標 (plDDT) 還可用於選擇準確的預測,以及區分強結合劑和弱結合劑。
研究以「Structure prediction of protein-ligand complexes from sequence information with Umol」為題,於2024 年5 月28 日發佈在《Nature Communications 》。

蛋白質與蛋白質標的接觸是評估新藥及重新定位已知物的重要議題。現有接觸方法有限制:需要高品質的蛋白質結構;難以確定準確的接觸姿態;多基於結合能力(親和力)評估,難以反映結構穩定性等其他因素。然而,現有的接觸方法限制在於需要高品質的蛋白質結構、準確的接觸姿態和多基於親和力評估。因此,採用蛋白質組合和結構評估相結合的方法,限制了對新配體的探索。
機器學習雖然已應用於此領域,但在針對已知標靶區域的表現上,仍未超越基於打分函數的經典方法。並且,預測的蛋白結構往往不適合直接用於配體對接。
此外,評估集中若結構基於發佈時間而非相似性劃分,會引入偏差,尤其是面對訓練中未見的受體結構時性能減半。
蛋白質靈活性對於達到結合狀態和成功對接至關重要,RoseTTAFold All-Atom 雖能在預測蛋白質時結合配體,其在 PoseBusters 測試集上的成功率也只有42%,且對未見過的蛋白質表現未知,顯示蛋白質-配體複合物結構預測的挑戰尚未完全解決。
柏林自由大學的團隊開發了一種 AI 方法,透過擴展 AlphaFold2 中的 EvoFormer,可以根據序列資訊預測蛋白質-配體複合物的結構。此網路與 RFAA 類似,不同之處在於不包括 3D 軌跡,使用模板結構或額外的晶體學配體資料作為輸入或在訓練期間使用。
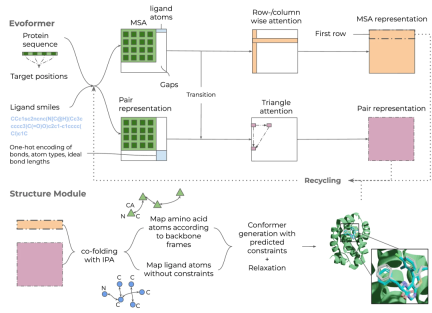
圖示:Umol 概述。 (來源:論文)
從蛋白質序列、可選蛋白質標靶(口袋)和配體 SMILES 開始,創建了多序列比對 (MSA) 和鍵矩陣。由此,在網路內產生特徵並產生 3D 結構。由於無需任何結構資訊即可產生最終的蛋白質-配體複合物結構,因此對蛋白質或配體的靈活性沒有任何限制。
與最接近的RoseTTAFold All-Atom 和NeuralPlexer1 相比,Umol 在PoseBusters 測試集上包含口袋資訊時獲得了更高的成功率(SR,配體RMSD ≤ 2 Å),分別為45 %、42%、24%,使其成為蛋白質-配體結構預測中表現最好的方法。

圖示:預測精度。 (資料來源:論文)
當從 Umol 中刪除口袋資訊並從 RFAA 中刪除模板資訊時,SR 分別下降到 18% 和 8%。當使用 AF 預測的 DiffDock 時,準確率為 21%,但取決於高度準確的介面預測(口袋 RMSD
許多略高於 2 Å 成功閾值的配體姿勢可能相當,這表明可能需要更靈活的評分系統。 Umol 在 2.35 Å 閾值下的成功率超過了 AutoDock Vina。在未使用天然蛋白質結構進行評分的情況下,即使是微小的對齊錯誤也會成為問題。
共折疊蛋白質-配體複合物具有加速藥物重新定位的潛力。特別是,研究人員發現配體的預測 lDDT (plDDT) 可用於選擇準確的對接姿勢,而蛋白質口袋的 pIDDT 適用於選擇準確的介面。

圖示:置信度指標與準確度。 (資料來源:論文)
配體 plDDT 也分離了高親和力配體和低親和力配體,這表明 Umol 和 Umol-pocket 不確定的一些預測可能是弱結合劑。這進一步證明了 Umol 的能力,並強調似乎已經了解了蛋白質-配體相互作用的重要方面。

圖示:BindingDB 預測。 (資料來源:論文)
儘管沒有口袋資訊的準確率為 18%,但網路仍可以在一定程度上區分強結合劑和弱結合劑。這對於註釋未知複合物特別有用,該團隊以非常高的置信度(配體 plDDT>85)呈現了 336 種蛋白質-配體結構。需要注意的是,雖然這些結構看似合理且其 L-plDDT 得分很高,但仍需透過實驗驗證。
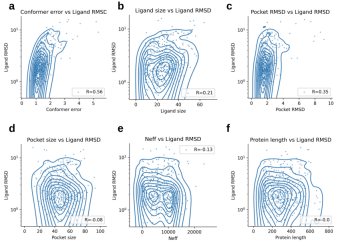
圖示:使用 Umol-pocket 分析 PoseBusters 測試集 (n=428) 上的預測的不同特徵與配體 RMSD (LRMSD) 之間的關係。 (資料來源:論文)
研究人員沒有發現模型的預測表現與「同蛋白質或配體相關的不同特徵」之間存在明確的關係。

圖示:最困難的 5 個結構。 (資料來源:論文)
然而,在其他方法難以預測的情況下,Umol-pocket 在 5 種情況下有 3 種是準確的。透過反轉訓練好的網絡,可以設計新的配體結合蛋白或蛋白質結合配體。另一種選擇是使用遷移學習來建立用於相同目的的生成擴散模型。在這種情況下,可以最大化配體或蛋白 plDDT 以嘗試創建高親和力結合物。
PDBbind 的目前版本包含 2019 年從 PDB 處理的資料。從那時起,已經提交了更多蛋白質-配體複合物,這表明可能可以實現更高的精度。
然而,目前尚不清楚需要什麼樣的精確度才能獲得有意義的蛋白質-配體對接結果。蛋白質結構預測的高精度在涉及其他分子(如小分子或 RNA)的任務中無法實現。
如果沒有蛋白質的共同演化訊息,結構預測的準確性會迅速下降。由於小分子或 RNA 沒有類似的資訊來源,因此人們只能依賴原子表徵。
表:PoseBuster 基準集上的成功率(配體 RMSD≤2Å 的百分比)除以 PDBBind 2020 版本的序列同一性 (seqid)。 (來源:論文)

研究人員認為口袋資訊非常有效,如果沒有口袋信息,深度學習方法似乎容易過度擬合。這項發現進一步證實了以下觀察結果:儘管 PoseBusters 測試集中的許多分子在訓練資料集中包含高度相似的類似物,但這種相似性與模型成功率無關。

圖示:一些測試。 (來源:論文)
對於基於結構的對接方法(如 Vina 或 Gold),未觀察到相同程度的過度擬合。這是意料之中的,因為它們是基於原子評分函數,因此不會在相同程度上依賴蛋白質同源性。
深度學習方法在訓練集上具有明顯更高的性能,這表明蛋白質同源性在蛋白質-配體對接中起著重要作用。 RFAA 在測試集上的表現高於訓練集,這表示訓練集和測試集之間可能存在資料外洩。
總之,要完全掌握蛋白質-配體相互作用的複雜性還有很長的路要走,但利用深度學習對整個複合物的結構進行預測可能會讓科學家更接近解決方案。
Umol:https://github.com/patrickbryant1/Umol
論文連結:https://www.nature.com/articles/s41467 -024-48837-6
以上是成功率超越RoseTTAFold系列,以序列資訊直接預測蛋白質-配體複合物結構的詳細內容。更多資訊請關注PHP中文網其他相關文章!

 加利福尼亞攻擊AI到快速賽道野火恢復許可證May 04, 2025 am 11:10 AM
加利福尼亞攻擊AI到快速賽道野火恢復許可證May 04, 2025 am 11:10 AMAI簡化了野火恢復允許 澳大利亞科技公司Archistar的AI軟件,利用機器學習和計算機視覺,可以自動評估建築計劃以符合當地法規。這種驗證前具有重要意義
 美國可以從愛沙尼亞AI驅動的數字政府中學到什麼May 04, 2025 am 11:09 AM
美國可以從愛沙尼亞AI驅動的數字政府中學到什麼May 04, 2025 am 11:09 AM愛沙尼亞的數字政府:美國的典範? 美國在官僚主義的效率低下方面掙扎,但愛沙尼亞提供了令人信服的選擇。 這個小國擁有由AI支持的近100%數字化的,以公民為中心的政府。 這不是
 通過生成AI的婚禮計劃May 04, 2025 am 11:08 AM
通過生成AI的婚禮計劃May 04, 2025 am 11:08 AM計劃婚禮是一項艱鉅的任務,即使是最有條理的夫婦,也常常壓倒了婚禮。 本文是關於AI影響的持續福布斯系列的一部分(請參閱此處的鏈接),探討了生成AI如何徹底改變婚禮計劃。 婚禮上
 什麼是數字防禦AI代理?May 04, 2025 am 11:07 AM
什麼是數字防禦AI代理?May 04, 2025 am 11:07 AM企業越來越多地利用AI代理商進行銷售,而政府則將其用於各種既定任務。 但是,消費者倡導強調個人需要擁有自己的AI代理人作為對經常定位的辯護的必要性
 商業領袖生成引擎優化指南(GEO)May 03, 2025 am 11:14 AM
商業領袖生成引擎優化指南(GEO)May 03, 2025 am 11:14 AMGoogle正在領導這一轉變。它的“ AI概述”功能已經為10億用戶提供服務,在任何人單擊鏈接之前提供完整的答案。 [^2] 其他球員也正在迅速獲得地面。 Chatgpt,Microsoft Copilot和PE
 該初創公司正在使用AI代理來與惡意廣告和模仿帳戶進行戰鬥May 03, 2025 am 11:13 AM
該初創公司正在使用AI代理來與惡意廣告和模仿帳戶進行戰鬥May 03, 2025 am 11:13 AM2022年,他創立了社會工程防禦初創公司Doppel,以此做到這一點。隨著網絡犯罪分子越來越高級的AI模型來渦輪增壓,Doppel的AI系統幫助企業對其進行了大規模的對抗 - 更快,更快,
 世界模型如何從根本上重塑生成AI和LLM的未來May 03, 2025 am 11:12 AM
世界模型如何從根本上重塑生成AI和LLM的未來May 03, 2025 am 11:12 AM瞧,通過與合適的世界模型進行交互,可以實質上提高生成的AI和LLM。 讓我們來談談。 對創新AI突破的這種分析是我正在進行的《福布斯》列的最新覆蓋範圍的一部分,包括
 2050年五月:我們要慶祝什麼?May 03, 2025 am 11:11 AM
2050年五月:我們要慶祝什麼?May 03, 2025 am 11:11 AM勞動節2050年。全國范圍內的公園充滿了享受傳統燒烤的家庭,而懷舊遊行則穿過城市街道。然而,慶祝活動現在具有像博物館般的品質 - 歷史重演而不是紀念C


熱AI工具

Undresser.AI Undress
人工智慧驅動的應用程序,用於創建逼真的裸體照片

AI Clothes Remover
用於從照片中去除衣服的線上人工智慧工具。

Undress AI Tool
免費脫衣圖片

Clothoff.io
AI脫衣器

Video Face Swap
使用我們完全免費的人工智慧換臉工具,輕鬆在任何影片中換臉!

熱門文章
熱工具

SublimeText3漢化版
中文版,非常好用

記事本++7.3.1
好用且免費的程式碼編輯器

禪工作室 13.0.1
強大的PHP整合開發環境

PhpStorm Mac 版本
最新(2018.2.1 )專業的PHP整合開發工具

SecLists
SecLists是最終安全測試人員的伙伴。它是一個包含各種類型清單的集合,這些清單在安全評估過程中經常使用,而且都在一個地方。 SecLists透過方便地提供安全測試人員可能需要的所有列表,幫助提高安全測試的效率和生產力。清單類型包括使用者名稱、密碼、URL、模糊測試有效載荷、敏感資料模式、Web shell等等。測試人員只需將此儲存庫拉到新的測試機上,他就可以存取所需的每種類型的清單。

