
計算效率提升100倍以上,上交李金金團隊開發以Transformer為基礎的大模型從頭計算分子動力學

- WBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWB原創
- 2024-06-18 18:02:541115瀏覽

作者 | 陶科豪
精確模擬原子與分子的動態行為對於開發新一代高效材料至關重要。
然而,傳統的從頭算分子動力學(AIMD)模擬自然提供了高精度的預測能力,但由於其高昂的計算成本和漫長的模擬時間,大大限制了研究的進度。
例如,建立一個包含100個原子的材料系統的30皮秒模擬通常需要數月時間,這對於需要快速迭代和優化的新材料研發構成了巨大挑戰。
在這種背景下,一個能夠顯著加快這一過程的人工智慧模型具有重要價值。
面對這些挑戰,上海交通大學人工智慧與微結構實驗室(AIMS-lab)開發了名為 T-AIMD 的革命性人工智慧模式。
這種模型採用了基於Transformer網路架構,不僅能夠高效降低計算成本,同時也能快速準確地預測任何離子在任何晶體結構中的行為。
透過這種方式,T-AIMD 模型將傳統的 AIMD 模擬速度提升了 100 倍以上,顯著加速了材料性能的評估過程。
此外,該模型還成功建構了一個龐大的混合離子導體資料庫,並在多項電池實驗中驗證了其預測的準確性。
此方法不僅在分子動力學模型(MD),生物藥物分子結合標靶、蛋白質摺疊、材料熱力學過程和機械性質計算等領域具有廣泛的應用潛力。
也為使用生成式人工智慧模型在更廣泛的科學領域內解決複雜問題提供了新的方法論。
T-AIMD 的成功應用展示了人工智慧技術在推動科學研究和技術創新中的巨大潛力,為未來的新材料研發和生物設計開發開闢了新的道路。
研究以「Transformer enables ion transport behavior evolution and conductivity regulation for solid electrolyte」為題,於 2024 年 6 月 11日發表在國際著名期刊上《國際著名期刊》(Yicegyage)。 論文的第一作者為上海交通大學人工智慧與微結構實驗室博士生陶科豪,通訊作者為實驗室主任李金金教授。
 文章連結:
文章連結:在人工智慧領域,Transformer 模式處理複雜序列資料的首選框架。
這種模型特別擅長從大規模資料中學習深層的模式和關聯,因此在語言處理、影像辨識以及各類預測任務中得到了廣泛應用。
儘管如此,在材料科學特別是從頭算分子動力學(AIMD)模擬的應用中, Transformer 的潛力尚未得到充分開發。
傳統的 AIMD 模擬在材料科學中非常重要,它能夠精確模擬原子和分子的動態行為。然而,這類模擬通常依賴重複的計算和昂貴的實驗,不僅耗時而且成本高昂。
面對這樣的挑戰,一個能夠快速提取和處理大量序列資料的智慧模型顯得特別重要。
針對這項需求,上海交通大學 AIMS-lab 團隊開發的 T-AIMD 模型,利用 Transformer 網路架構,顯著提升了 AIMD 模擬的速度和準確性。
這種新型模型能夠在大幅降低計算成本的同時,快速且準確地分析和預測原子及分子在各種條件下的行為。
與傳統 AIMD 方法相比,T-AIMD 能將模擬速度提高 100 倍以上,同時保持了預測的高準確性,大幅縮短了材料研發的周期。
這不僅為材料科學領域的研究提供了新的工具,也展示了 AI 在高效能運算任務中的應用潛力,為未來的科學探索開闢了新的可能。
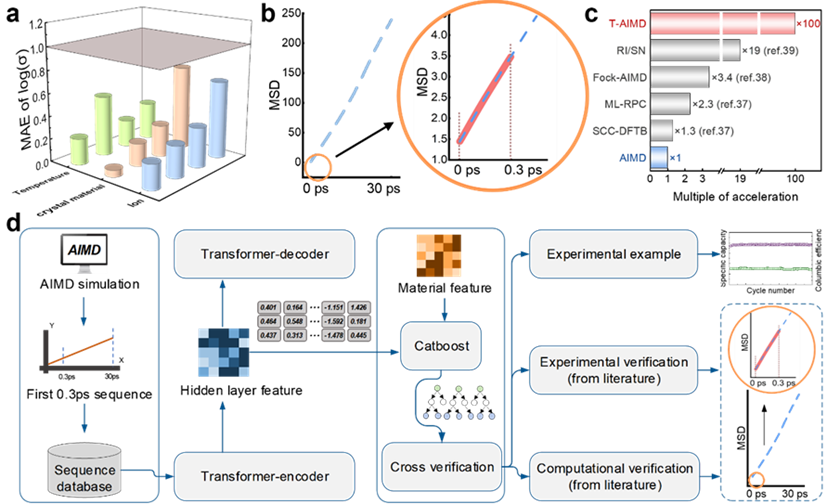 圖示:T-AIMD 預測結果示意及工作流程示意圖。 (資料來源:論文)
圖示:T-AIMD 預測結果示意及工作流程示意圖。 (資料來源:論文)
以解決固態電解質中離子輸運行為的預測問題為例。該模型透過學習離子在電解質中的擴散序列,能夠預測其在未來狀態下的行為,大大加速了材料性能的評估過程。
此外,T-AIMD 模型還結合了多源材料描述符,增強了其在處理複雜材料系統中的應用能力,使其不僅能預測單一離子種類的行為,還能處理多離子系統中的交互作用和複雜動力學問題。
這種基於 Transformer 的新方法為固態電解質的研發提供了一種全新的視角和工具,有望在材料科學領域開創新的研究和應用前景。
關於 T-AIMD 如何運作
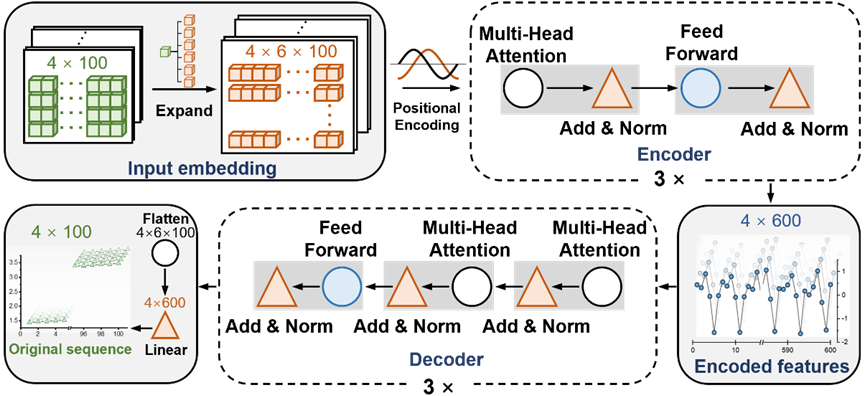
圖示:T-AIMD 的網路架構圖。 (資料來源:論文)
T-AIMD(Transformer-based Ab Initio Molecular Dynamics)是一種結合了從頭算分子動力學(AIMD)模擬和Transformer 深度學習架構的模型,旨在提高固態電解質材料中離子輸運特性的預測速度和準確度。這種模型的工作原理可以分為以下幾個關鍵步驟:
1、 資料準備和預處理
T-AIMD 首先收集材料的離子擴散數據,這些數據來自於傳統的AIMD模擬。這些模擬產生的數據包括時間序列數據,記錄了離子在電解質中的移動軌跡。對這些序列資料進行預處理,將其轉換為適合機器學習模型輸入的格式。
2、 特徵提取
利用 Transformer 模型的編碼器部分,T-AIMD 能夠從序列資料中提取關鍵特徵。在這個過程中,模型透過自註意力機制捕捉序列中的長距離依賴關係,這對於理解複雜的離子動力學非常關鍵。
3、 序列學習和預測
在特徵提取後,Transformer 模型的解碼器部分被用來基於已編碼的特徵進行序列預測。在這一步驟中,模型不僅可以預測離子的未來行為,還可以分析在不同條件下(如不同溫度和壓力)離子的潛在行為。此外,模型透過這些學習到的特徵,能夠預測材料的離子導電性等關鍵性能指標。
4、多源材料描述符的整合
T-AIMD 結合了來自不同來源的材料描述符,如晶體結構、離子種類和電子屬性等,這有助於模型更全面地理解和預測材料性能。這種整合方法提高了模型在不同材料系統中的通用性和適應性。
5、 模型驗證和應用
開發完成的模型需要透過與實驗數據和其他計算方法的比較來驗證其預測準確性。驗證成功後,T-AIMD 可以用於快速篩選和優化新的目標材料,大幅縮短研發週期並降低成本。
關於T-AIMD 的穩健性能
T-AIMD 模型的穩健性能主要表現在以下幾個方面:
1、準確性
T-AIMD 模型整合了Transformer 架構,極大化AIMD 架構了其學習和預測複雜動力學行為的能力。在 AIMD 模擬加速方面,T-AIMD 顯示出比傳統方法更高的準確性。這得歸功於深度學習技術的應用,使模型能夠在更短的時間內精確預測更長尺度的離子行為。
2、計算效率
在計算效率方面,T-AIMD 顯著優於傳統 AIMD 方法。傳統的 AIMD 模擬需耗費大量時間模擬離子擴散,而 T-AIMD 透過優化運算流程,顯著降低了對高效能運算資源的依賴,將模擬時間從幾個月縮短至幾天或幾小時。
3、通用性和靈活性
T-AIMD 能處理比傳統機器學習模型(如支援向量機或決策樹)更複雜的資料結構和更大的資料集。該模型能夠適應多種類型的材料,並有效預測在不同環境條件(如溫度和壓力的變化)下的行為。
4、模型穩健性
T-AIMD 在處理帶有雜訊和不確定性的資料時表現出較高的穩健性。在對比實驗中,即使在數據存在輕微偏差的情況下,T-AIMD 也能保持高度的預測精度,這是其他簡單機器學習模型難以達到的。
5、擴展性和適應性
T-AIMD 模型的架構允許靈活的調整和優化,適應不斷變化的研究需求和新科學發現。這種擴展性使得 T-AIMD 在未來研究中能夠持續發揮關鍵作用,其應用不限於固態電解質,還可拓展到其他能源材料和複雜化學系統的研究。
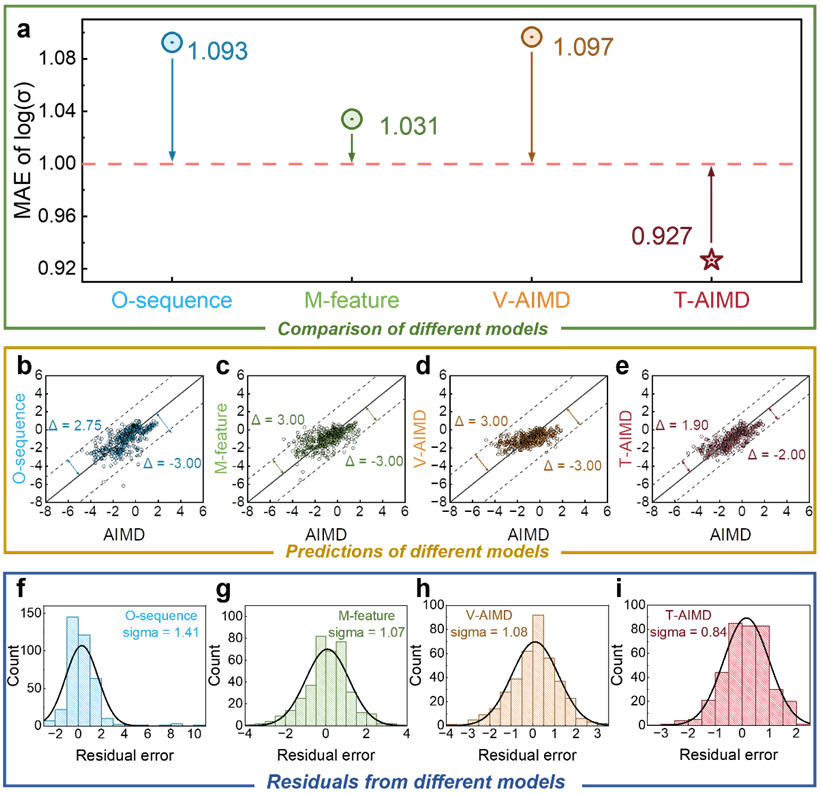
綜上所述,基於T-AIMD 框架,可大幅加快分子動力學的模擬效率,提升效率1000 倍、10000 倍、甚至更多,為材料製造和生物設計節省大量的時間成本。
T-AIMD 模型在多個關鍵方面均優於傳統 AIMD 模擬和其他機器學習方法,文本給出的例子顯示了其在固態電解質研究和開發中的強大潛力和應用前景。
T-AIMD 的實用性遠不止於此。該模型的強大功能和靈活性使其能廣泛應用於材料科學的多個領域。
未來,它有望可以用於預測其他類型材料如半導體、金屬和高分子材料中的離子和分子行為。
此外,T-AIMD 模型的能力不限於單一離子種類的行為預測,它還能處理多離子系統中的複雜相互作用和動力學問題,這使得它在設計新材料和改善現有材料的性能方面具有極高的實用價值。
以上是計算效率提升100倍以上,上交李金金團隊開發以Transformer為基礎的大模型從頭計算分子動力學的詳細內容。更多資訊請關注PHP中文網其他相關文章!