
UW-중국 팀의 새로운 다중 모드 데이터 분석 및 생성 방법 JAMIE는 세포 유형 및 기능 예측 능력을 크게 향상시킵니다.

- WBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWB앞으로
- 2023-06-10 14:43:291499검색
최근에는 단세포 기술의 급속한 발전으로 단세포의 다양한 특성을 측정하여 단세포 다중 모드 데이터(예: scRNA-seq, scATAC-seq, Patch-seq)를 얻을 수 있게 되었습니다. ).
이 데이터는 세포 기능과 분자 메커니즘을 더 깊이 이해하는 데 도움이 됩니다. 예를 들어, 연구자들은 최근 기계 학습 방법을 사용하여 단일 세포 다중 모드 데이터 간의 관계를 분석하여 세포 유형 및 질병과 관련된 생물학적 메커니즘을 이해했습니다.
그러나 단일 셀 다중 모드 데이터를 획득하는 데는 비용이 많이 들고 모드 손실이 자주 발생합니다. 기존 기계 학습 방법은 일반적으로 데이터 채우기 및 삽입을 위해 완전히 일치하는 다중 모드 데이터가 필요하며 양식이 누락된 상황에는 적합하지 않습니다.
이 문제를 해결하기 위해 University of Wisconsin-Madison의 Wang Daifeng 연구실에서는 JAMIE(Joint Variational Autoencoders for Multimodal Imputation and Embedding)라는 공동 변형 자동 인코더를 기반으로 한 오픈 소스 기계 학습 방법을 개발했습니다.
JAMIE는 세포 유형과 기능을 더 잘 예측하기 위해 데이터 정렬, 임베딩, 누락 데이터 보완 등 단일 세포 다중 모드 데이터의 통합 분석에 사용할 수 있습니다.
이 연구는 최근 Nature Machine Intelligence에 게재되었습니다.

논문 주소: https://www.nature.com/articles/s42256-023-00663-z
프로젝트 주소: https://github.com/daifengwanglab /JAMIE
JAMIE 방법 소개
JAMIE는 재사용 가능한 관절 변형 자동 인코더 모델을 훈련하여 사용 가능한 다중 모드 데이터를 유사한 잠재 공간에 별도로 투영함으로써 단일 모드를 향상시킵니다. 상태 패턴을 추론하는 능력입니다.
그림 1에 표시된 것처럼 교차 모달 대체를 수행하기 위해 JAMIE는 데이터를 인코더에 공급한 다음 반대 디코더를 통해 잠재 공간 결과를 처리합니다.
JAMIE는 자동 인코더의 재사용 가능하고 유연한 잠재 공간 생성과 정렬 방법의 자동 대응 추정을 결합하여 불완전한 대응이 있는 다중 모드 데이터 처리를 가능하게 합니다.
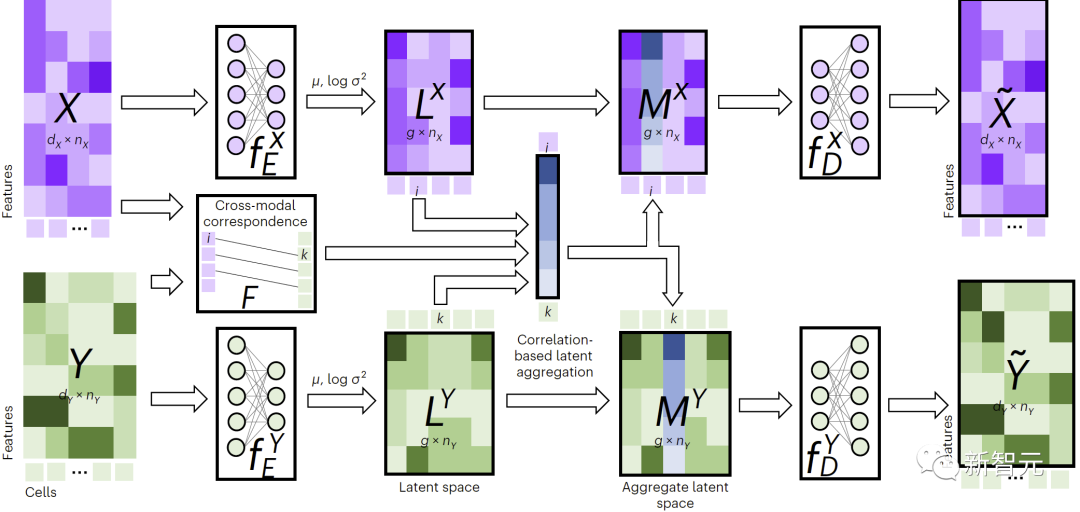
그림 1. JAMIE 방법 개요
구체적으로 JAMIE는 다음 두 단계로 나눌 수 있습니다.
- 입력 데이터 전처리. 바이모달 모드를 예로 들어, 모드에 해당하는 데이터 행렬은 각각 과 이라고 가정합니다. 여기서는 특징 차원과 합계가 다를 수 있으며 샘플 수도 다를 수 있습니다. 전처리는 각 행렬의 각 행을 정규화하여 평균이 0이고 분산이 1이 되도록 합니다. 해당 데이터가 있는 경우 사용자는 모달 상관 행렬을 제공하여 성능을 향상시킬 수 있습니다. 여기서는 모달의 6번째 샘플이 모달의 7번째 샘플과 완전히 일치함을 의미하고 알려진 일치성이 없음을 의미하며 부분적인 부분이 있음을 의미합니다. 서신.
- 관절 변형 자동 인코더를 사용하여 각 양식의 유사성 잠재 공간을 학습합니다. 여기서 (기본값, 사용자 조정 가능)는 잠재 공간 차원입니다. 훈련 과정에서 JAMIE는 다음 손실 함수를 최소화합니다.
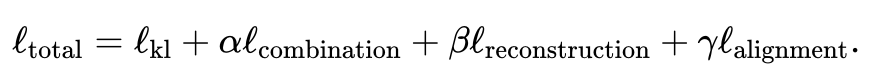
총 손실 함수에는 4개의 항목이 포함됩니다.
첫 번째 항목은 변이 자동 인코더에서 추론한 분포와 다변량 표준 정규 분포 간의 Kullback-Leibler(KL) 발산을 계산합니다. 이는 잠재 공간의 연속성을 유지하는 데 도움이 됩니다. 해당 샘플은 재구성된 데이터 행렬과 원본 데이터 행렬 사이의 평균 제곱 오차의 합입니다. 네 번째 항은 생성된 잠재 공간을 조정하기 위해 추론된 교차 모드 대응을 사용합니다.
각 항목의 구체적인 표현은 논문 원문을 참고해주세요. 첫 번째 항목에 대한 두 번째, 세 번째, 네 번째 항목의 가중치는 사용자가 조정할 수 있으며, JAMIE는 일반적인 상황에 적합한 기본 가중치도 제공합니다.
다음 표는 JAMIE의 모델과 적용 범위를 현재의 최신 방법과 비교한 것입니다. JAMIE는 여러 가지 통합 및 보간 방법의 기능을 단일 아키텍처로 통합함으로써 누락된 양식 보간이 가능하고 비오믹스 데이터 호환성이 가능하며 부분적인 대응만으로 다중 모드 데이터를 처리할 수 있는 기능이 장점입니다.
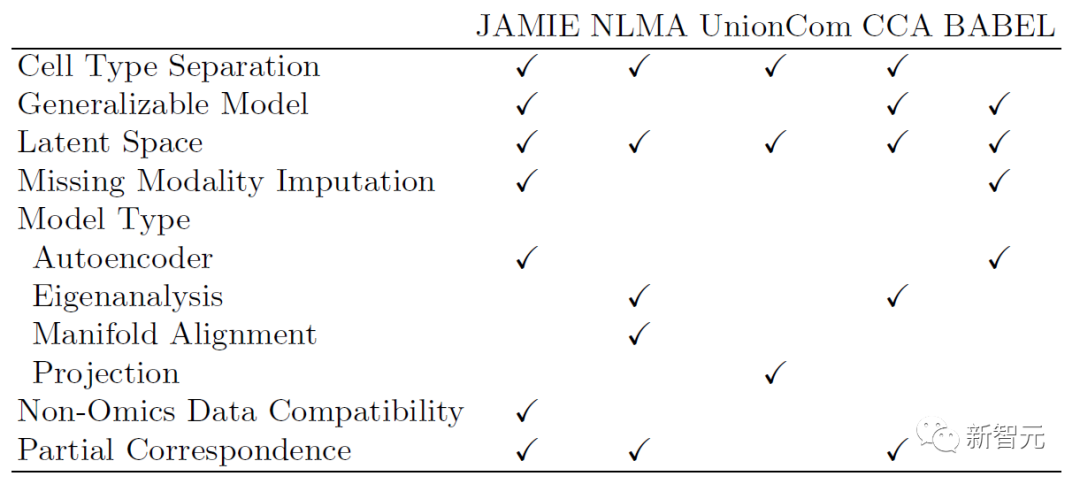
표 1. 다양한 다중 모달 통합 및 누락된 모달 채우기 방법 비교. 단일 아키텍처를 통해 JAMIE는 다양한 통합 및 보간 방법의 기능을 통합합니다. NLMA: 비선형 다양체 정렬 [15], UnionCom [7], CCA: 정규 상관 분석 [15, 16], BABEL [5].
JAMIE의 주요 애플리케이션
다중 모드 데이터의 통합 및 표현형 예측
다중 모드 데이터의 통합은 분류 성능을 향상시키고 표현형 지식을 향상시키며 복잡한 생물학적 메커니즘에 대한 이해를 향상시킬 수 있습니다.
두 개의 데이터 세트와 해당 관계가 주어지면 JAMIE는 훈련된 인코더와 을 기반으로 잠재 공간 데이터를 생성하고 을 기반으로 클러스터링 또는 분류를 수행할 수 있습니다.
잠재 공간 데이터를 기반으로 한 클러스터링에는 두 가지 방식을 기능 생성에 통합하는 등 여러 가지 장점이 있습니다. 그런 다음 JAMIE는 세포 유형 예측과 같은 샘플 대응을 예측할 수 있습니다.
부분적으로 레이블이 지정된 데이터 세트의 경우 동일한 클러스터의 셀은 유사한 유형을 가져야 합니다.
JAMIE는 잠재 공간 데이터를 생성하는 과정에서 다양한 유형의 데이터 특성을 분리하므로 일반적으로 더 나은 결과를 얻기 위해 복잡한 클러스터링이나 분류 알고리즘이 필요하지 않습니다.
고차원 데이터의 경우 JAMIE는 세포 유형 클러스터링 시각화를 위해 UMAP [32]를 사용합니다.
교차 모드 데이터 대체
현재의 많은 교차 모드 대체 방법은 대체 목적을 위한 기본 생물학적 메커니즘을 학습했음을 입증할 수 없습니다.
피드포워드 네트워크나 선형 회귀 방법과 비교할 때 JAMIE는 기본 생물학적 메커니즘을 더 잘 학습하여 보다 엄격한 수학적 기초를 기반으로 누락된 데이터를 예측할 수 있습니다.
그림 2는 교차 모달 데이터 채우기를 위한 JAMIE의 프로세스를 보여줍니다. JAMIE는 먼저 훈련 데이터에 대한 인코딩 및 디코딩 모델을 훈련합니다.
새로운 데이터에 대해 JAMIE는 먼저 데이터에서 학습한 인코더를 사용하여 이를 잠재 공간에 투영하여 얻은 다음 잠재 공간 특징을 집계하여 얻은 다음 마지막으로 해당 디코더를 통해 누락된 패턴 데이터로 디코딩합니다.
JAMIE는 잠재 공간을 사용하여 세포 간의 일치성을 예측하며, 이는 데이터 특징과 표현형 간의 관계를 이해하는 데 도움이 될 수 있습니다.
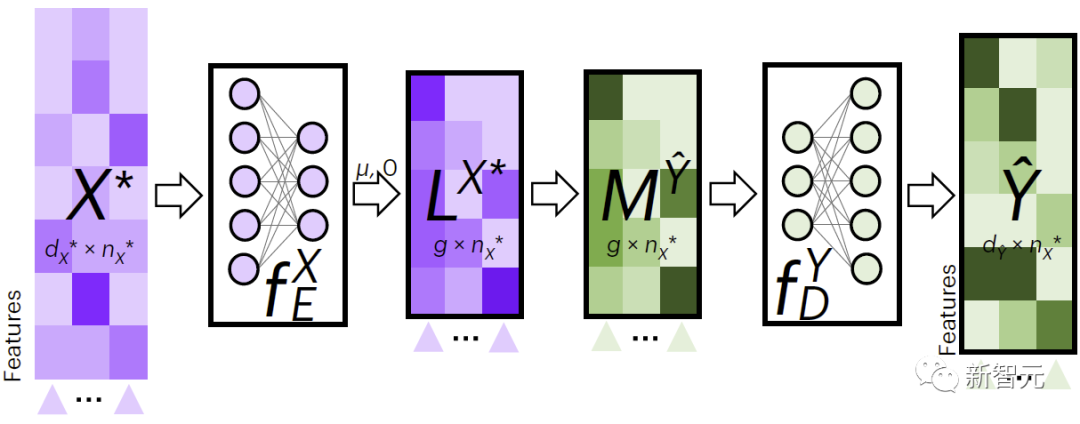
그림 2. JAMIE 교차 모달 보간
잠재 공간 기능 및 채우기 기능 설명
훈련된 모델인 JAMIE는 SHAP(SHapley)를 채택했습니다. 부가적인 설명)[18].
SHAP은 모델에서 생성된 개별 예측을 샘플 변조하여 개별 입력 기능의 중요성을 평가합니다. 이는 다양하고 흥미로운 응용 분야에 사용될 수 있습니다.
대상 변수를 표현형별로 쉽게 분리할 수 있다면 SHAP는 추가 연구를 위해 관련 특성을 식별할 수 있습니다. 또한, 대치(imputation)를 수행하면 SHAP는 모델이 학습한 모달 간 연결을 드러낼 수 있습니다.
모델과 샘플이 주어지면 배경 특징 벡터가 어디에 있는지와 같은 SHAP 값을 학습합니다.
이면 SHAP 값과 배경 출력의 합은 와 같으며, 여기서 각각은 모델 출력에 미치는 영향에 비례합니다.
또 다른 유용한 기술은 분류(예: LTA [7, 19]) 또는 대치(예: 대치된 특징과 측정된 특징 간의 대응)를 위한 핵심 메트릭을 선택하고 이를 모델에서 하나씩 사용하는 것입니다. 메트릭은 다음과 같습니다. 각 기능을 배경 값으로 제거(교체)하여 평가합니다.
그런 다음 주요 측정항목이 악화되면 제거된 기능이 모델 결과에 더 중요하다는 것을 나타냅니다.
실험 결과
JAMIE는 검증을 위해 일반적으로 사용되는 4개의 단일 셀 다중 모드 데이터 세트를 사용했습니다.
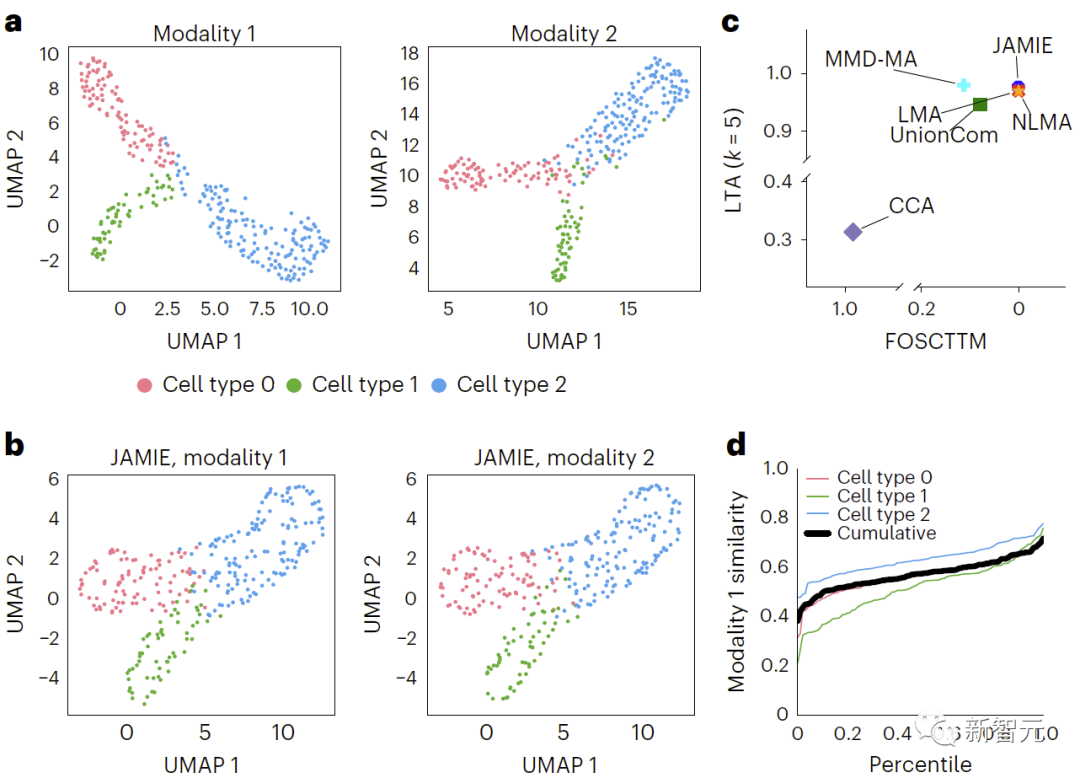
(1) MMD-MA에서 분기된 다양체의 가우스 분포 샘플링으로 생성된 시뮬레이션된 다중 모달 데이터(300개 샘플, 3개 세포 유형)
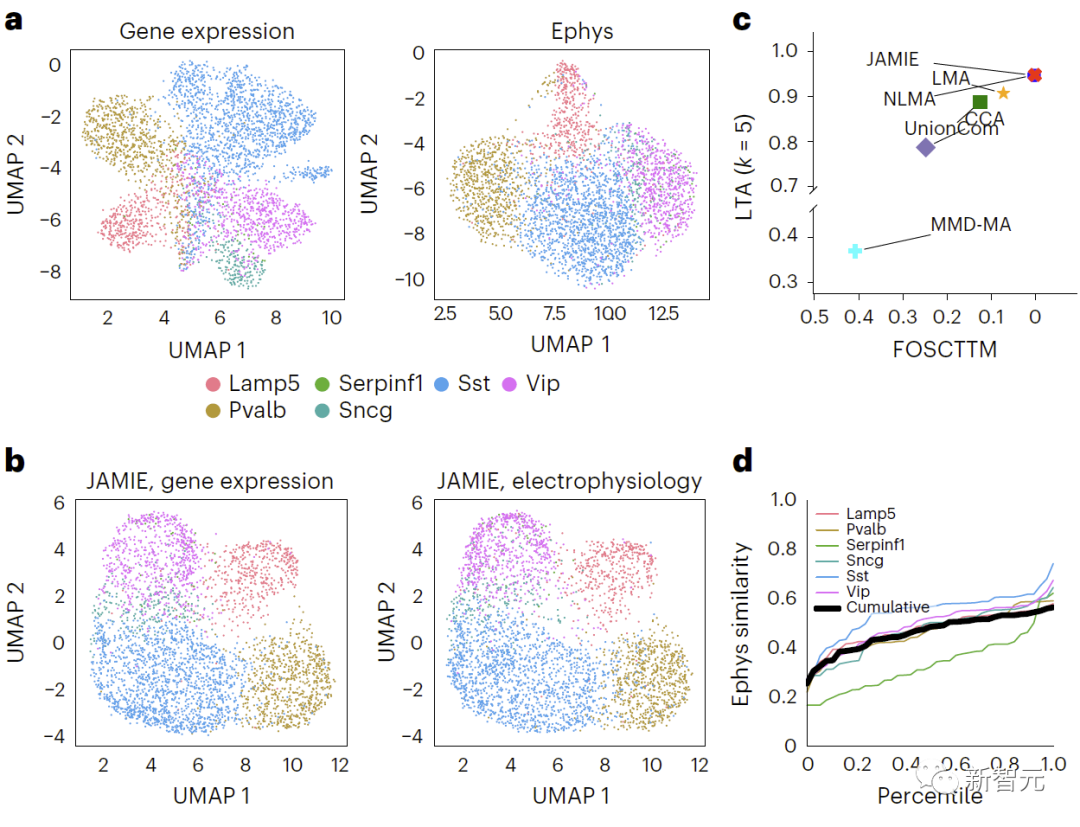
(2) 마우스 시각 피질(패치-seq 유전자 발현 및 전기생리학적) 10x 단일 세포 다중 오믹스 유전자 발현 및 염색질에서 얻은 마우스 운동 피질(1,208개 샘플, 9개 세포 유형) 및 마우스 운동 피질(1,208개 샘플, 9개 세포 유형)의 단일 뉴런 세포 특성화 데이터 인간 발달 중인 뇌의 8,981개 샘플에 대한 접근성 데이터(임신 21주, 인간 대뇌 피질의 7개 주요 세포 유형 포함)
(4) COLO-320DM 결장 선암종 세포주.
평가 결과 JAMIE가 다른 방법보다 훨씬 우수하고(그림 3의 MMD-MA 가지 다양체 시뮬레이션 데이터 결과 비교, 그림 4의 마우스 시각 피질 데이터 결과 비교) 다중 모드를 우선시하는 것으로 나타났습니다. 셀룰러 해상도에서 잠재적으로 새로운 기계적 통찰력을 제공하는 동시에 중요한 기능을 채웁니다.
요약
요약하자면 JAMIE는 단일 세포 다중 모드 데이터의 통합 예측을 위한 새로운 심층 신경망 모델입니다.관절 변형 자동 인코더(VAE) 구조에 의존하는 새로운 잠재 임베딩 집계 방법을 통해 구현된 복잡하거나 혼합되거나 부분적으로 대응하는 다중 모드 데이터에 적합합니다. 위에서 언급한 뛰어난 성능 외에도 JAMIE는 효율적인 컴퓨팅 기능과 낮은 메모리 사용량 요구 사항도 갖추고 있습니다. 또한 사전 훈련된 모델과 학습된 교차 모달 잠재 임베딩을 다운스트림 분석에 재사용할 수 있습니다.
물론 더 큰 데이터 세트의 경우 VAE(변형 자동 인코더)를 훈련하는 데 많은 시간이 걸립니다. 따라서 JAMIE의 자동 PCA와 같은 이전 기능 선택 방법은 시간 요구 사항을 완화하는 데 도움이 됩니다. VAE는 재구성 손실을 사용하므로 크거나 반복되는 특징이 저차원 임베디드 특징에 불균형적으로 영향을 미치는 것을 방지하기 위해 데이터 전처리도 중요합니다. 특정 교차 모달 대체의 경우 최종 모델의 편향과 일반화 능력에 부정적인 영향을 미치지 않도록 훈련 데이터 세트의 다양성을 신중하게 고려해야 합니다. JAMIE는 잠재적으로 확장되어 다양한 조건의 유전자 발현 데이터와 같은 다양한 양식이 아닌 다양한 소스의 데이터 세트를 정렬할 수도 있습니다.
저자 소개
논문의 저자는 Noah Cohen Kalafut(컴퓨터과학과 박사과정 학생), Huang Xiang(선임연구원), Wang Daifeng(PI)입니다. 위스콘신대학교 매디슨 캠퍼스 생물통계학과, 의료정보학과, 컴퓨터공학과, 와이즈만 연구센터. 교신저자는 Wang Daifeng 교수이다.
1973년에 설립된 와이즈만 센터는 반세기 동안 인간 발달, 신경 발달 장애 및 신경 퇴행성 질환에 대한 연구를 발전시켜 왔습니다.
위 내용은 UW-중국 팀의 새로운 다중 모드 데이터 분석 및 생성 방법 JAMIE는 세포 유형 및 기능 예측 능력을 크게 향상시킵니다.의 상세 내용입니다. 자세한 내용은 PHP 중국어 웹사이트의 기타 관련 기사를 참조하세요!