
게놈 형태 예측 모델 및 고처리량 컴퓨터 유전자 스크리닝 방법 탐색 및 응용

- PHPz앞으로
- 2023-05-08 14:16:08885검색

그림 0
다양한 유형의 세포에서 게놈 형태의 차이가 유전자 발현의 특이성을 결정하며, 이는 결국 다양한 세포 유형의 기능적 차이를 결정합니다. 오랫동안 in situ 하이브리드화부터 Hi-C 및 micro-C 기술과 같은 고처리량 검출에 이르기까지 게놈 형태 검출을 위한 실험 방법은 일반적으로 시간이 많이 걸리고 노동 집약적이며 비용이 많이 들고 강력한 기술적 한계가 있습니다. 이러한 방법은 게놈 형태 연구 분야, 특히 희귀 세포 유형 연구 및 대규모 게놈 형태 조절의 인과 관계를 검증할 필요성에서 이러한 실험 기술의 광범위한 적용을 크게 제한합니다. 이러한 방법의 한계로 인해 오랫동안 3차원 게놈 구조 조절 분야의 새로운 발견이 제한되었습니다.

그림 1
2023년 1월 9일 Aristotelis Tsirigos 연구소와 Broad Institute of MIT 및 Harvard, NYU Grossman School of Medicine Xia Bo의 연구실이 협력하여 "세포 유형별 특정 세포 3D 염색질 조직의 예측은 Nature Biotechnology에서 실리코 유전자 스크리닝에서 높은 처리량을 가능하게 합니다.

논문 주소: https://www.nature.com/articles/s41587-022-01612-8
본 연구에서는 Ph.D., New York University School 의학부 Sheng Tan Jimin과 Xia Bo 박사는 특정 세포 유형의 염색질 구조를 예측하기 위해 새로운 다중 모드 기계 학습 모델 C.Origami를 최초로 제안했으며, 새로운 고처리량 컴퓨터 유전 스크리닝(in silico 유전 스크리닝) 기반을 제안했습니다. , ISGS) 방법을 사용하여 세포 유형별 기능적 게놈 요소를 식별하고 새로운 염색질 구조 조절 메커니즘을 발견하는 데 사용됩니다.
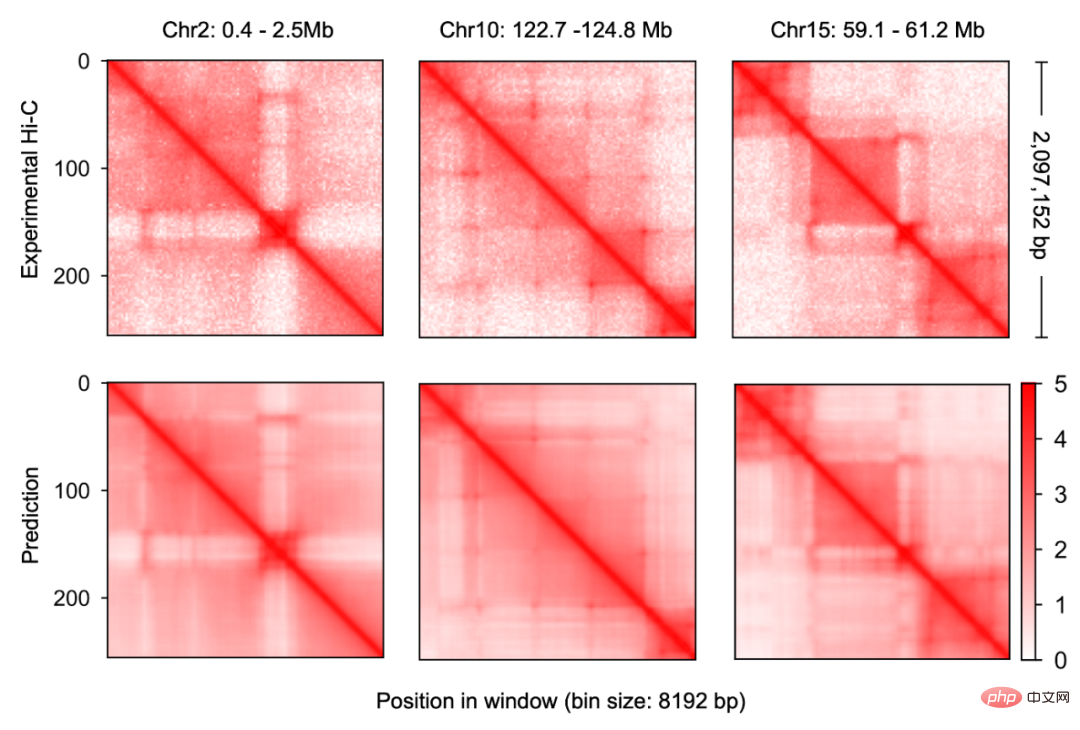
그림 2
연구원 먼저 게놈 데이터를 위한 새로운 다중 모드 딥 러닝 프레임워크인 Origami를 구축하여 DNA 서열 정보와 세포를 효과적으로 통합할 수 있습니다. 특정 기능 게놈 정보를 수집하고 새로운 게놈 정보를 예측합니다. 연구진은 반복적인 디버깅과 모델 훈련을 통해 DNA 서열, CTCF 결합 상태(CTCF ChIP-seq), ATAC-seq 신호를 입력 정보로 통합하면 염색질 형태를 정확하게 예측할 수 있으며, 2차원 Hi-C 매트릭스를 다음과 같이 사용할 수 있음을 발견했습니다. 출력 목표를 예측합니다(그림 1-2). 입력정보는 200만 염기쌍의 DNA, CTCF ChIP-seq, ATAC-seq이었다. 연구자들은 Onehot 인코딩을 사용하여 개별 DNA 서열을 인코딩하는 반면, CTCF ChIP-seq 및 ATAC-seq는 개별적이지 않은 특징을 인코딩합니다.

C.Origami 모델은 DNA와 게놈 정보를 처리하고 압축하는 인코더, Transformer 중간 계층 및 출력 Hi-C 디코더 의 세 부분으로 나뉩니다. 인코더는 일련의 1D ResNet과 strided convolution으로 구성되어 200만 염기쌍의 입력 정보를 인코딩하고 압축합니다. 인코더 끝에서 200만 길이의 메시지는 256 길이로 압축되어 변환기에 대한 입력 메시지로 사용됩니다. Transformer의 self-attention 메커니즘은 서로 다른 게놈 영역 간의 상호 의존성을 처리하고 모델의 전반적인 성능을 향상시킬 수 있습니다. Transformer의 어텐션 매트릭스는 모델의 해석 가능성을 향상시킬 수도 있습니다. 연구원들은 예측 시 다양한 영역에 대한 모델의 강조점을 측정하기 위해 주의 가중치를 "주의 점수"로 변환했습니다. 마지막으로 연구진은 Hi-C 디코더의 입력 정보로 사용된 "외부 연결"을 사용하여 Transformer 모듈의 1D 출력을 2D 접촉/인접 행렬로 변환했습니다. 디코더는 Dilated 2D ResNet입니다. 연구진은 최종 레이어의 각 픽셀 위치에 있는 수용 필드가 모든 입력 정보를 포괄할 수 있도록 다양한 레이어의 확장 인자를 조정했습니다.
염색질 형태를 예측하는 이 모델을 C.Origami라고 합니다. 연구원들은 C.Origami를 유전체학 분야 최초의 다중 모드 딥 러닝 모델이라고 부릅니다. 다중 모드 특성으로 인해 C.Origami는 노출된 적이 없는 새로운 세포 유형의 염색질 형태를 정확하게 예측(새로운 예측)할 수 있습니다. 예를 들어, IMR-90 세포(폐 섬유아세포)에 대해 훈련된 모델은 GM12878 세포(B 림프구)의 특정 염색질 형태를 정확하게 예측할 수 있었습니다(그림 3).

그림 3
구조적 변형(예: 염색체 전좌)은 종양에서 매우 흔하며 종종 염색질 상호 작용 패턴을 변경하여 종양 유전자 또는 종양 억제 유전자의 발현에 영향을 미칠 수 있습니다. 염색질 형태와 유전자 발현에 대한 이러한 구조적 변화의 영향을 연구하는 것은 종양 발생 및 진행 메커니즘을 이해하는 데 중요합니다. 이러한 유형의 연구는 일반적으로 구조적 변이 부위의 염색질 형태를 분석하기 위해 4C-seq 또는 Hi-C와 같은 실험을 사용해야 하지만, 자원과 시간의 제약이 있고 대규모로 수행하기 어려운 경우가 많습니다.
이 연구에서 C.Origami는 입력 변수의 DNA 서열 변화를 시뮬레이션한 다음 돌연변이된 암 게놈에서 새로운 염색질 상호 작용을 예측할 수 있습니다. 이전 연구에서는 T 세포 급성 림프구성 백혈병(T-ALL) 세포 모델 CUTLL1에 chr7-chr9 염색체 전좌가 있는 것으로 나타났습니다(그림 4). C. Origami는 염색체 전좌 변형을 계산적으로 시뮬레이션함으로써 변형 부위에서 새로운 TAD 구조를 정확하게 예측하고 chr9에서 chr7까지 확장되는 '염색질 줄무늬' 구조를 감지했습니다(그림 4).

그림 4
C.Origami의 정확한 예측 효과를 고려하고 역유전자 선별 원리에서 영감을 받아 연구진은 새로운 고처리량 전산 유전자 선별(in silico)을 제안했습니다. 유전자 스크리닝, ISGS) 방법을 사용하여 세포 유형별 기능성 게놈 요소를 체계적으로 식별하고 새로운 염색 조절 분자를 발견하는 데 도움을 줍니다(그림 5). 연구자들은 염색질 구조에 필요한 cis-조절 요소를 체계적으로 식별하기 위해 C. Origami 모델을 기반으로 컴퓨터 유전 스크리닝 ISGS를 위한 프레임워크를 개발했습니다. 게놈 전체의 1kb 해상도 ISGS를 사용하여 저자는 염색질 형태에 중요한 영향을 미치는 cis 조절 요소(게놈의 ~1%)를 분리했습니다. 이러한 염색질 형태 조절 서열은 CTCF 결합 및 ATAC-seq 신호에 대해 차별적인 의존성을 나타냅니다(그림 5).

사진 5
ISGS 프레임워크는 세포 또는 질병 특이적 염색질 형태에 대한 높은 처리량 스크리닝을 가능하게 합니다. 연구진은 CUTLL1, Jurkat 및 정상 T 세포에서 ISGS를 수행한 결과 CHD4 유전자 근처의 시스 조절 요소(CHD4-insu)가 T-ALL 세포에서 특이적으로 손실된다는 사실을 발견했습니다. 스크리닝 결과는 T-ALL 세포에서 CHD4-insu의 절연 손실로 인해 CHD4 유전자가 새로운 염색질 상호 작용을 확립하여 CHD4 발현을 상향 조절하고 백혈병 세포 증식을 촉진할 수 있음을 나타냅니다.
ISGS는 염색질 형태를 조절하는 새로운 거래 인자를 체계적으로 발견하는 데에도 사용될 수 있습니다. 중요한 세포 유형별 조절 서열과 전사 인자 결합 부위에 대한 농축 분석을 통해 연구진은 세포 유형별 게놈 형태에 기여하는 조절 인자를 확인했습니다. 흥미롭게도 이전 연구에서는 MAZ가 CTCF와 함께 염색질 형태를 조절할 수 있음을 발견했습니다. ISGS 및 전사 인자 농축 분석을 통해 저자는 MAZ가 개방형 염색질 영역에서 크게 농축된 반면, CTCF가 결합하는 비개방형 염색질 영역에서는 약한 결합만 나타냄을 발견했습니다. 이 결과는 MAZ가 CTCF와 독립적으로 게놈 형태를 조절할 수 있음을 시사합니다.
연구원들은 염색질 구조 예측에서 DNA 서열과 염색질 정보를 결합하는 다중 모드 기계 학습 모델의 큰 잠재력을 확인했습니다. Origami 모델의 기본 다중 모드 아키텍처는 후생적 변형, 유전자 발현, 돌연변이의 기능적 스크리닝 등과 같은 다른 유전체학 데이터의 적용으로 확장될 수 있습니다. 연구자들은 미래의 유전체학 연구가 생물학적 실험을 통해 검증된 차세대 고처리량 연구 방법으로 보완된 1차 전산 유전자 스크리닝 도구로서 딥 러닝 모델을 사용하는 방향으로 더 많이 전환될 것이라고 예측합니다.
본 연구에는 뉴욕대학교 의과대학 박사과정생인 Tan Jimin이 제1저자로, Aristotelis Tsirigos 박사와 Xia Bo 박사가 공동교신저자로 참여했습니다. 이 연구는 2020년 10월 전염병 봉쇄 기간 동안 Xia Bo와 Tan Jimin의 브레인스토밍으로 시작되었습니다. 2년 반 동안의 개선과 다듬질 끝에 2023년 1월 Nature Biotechnology에 공식적으로 게재되었습니다.
본 프로젝트의 코드와 학습 데이터는 GitHub와 Zenodo에 오픈소스로 공개되어 있으며, 기능 시연을 위해 Google Colab을 탑재하고 있습니다.
프로젝트 주소: https://github.com/tanjimin/C.Origami
교신저자
Dr. Xia Bo’s Laboratory (Broad Institute of MIT and Harvard) 홈페이지: www.boxialab.org
Xia Bo 박사는 게놈의 3차원 형태를 조절하는 핵심 메커니즘과 인간 질병, 발달 및 진화에 대한 생물학적 중요성을 분석하는 데 전념하고 있습니다. Xia Bo의 연구실에서는 같은 생각을 가진 박사후 연구원이 팀에 합류하는 것을 환영합니다.
Tsirigos Lab(뉴욕대학교 Grossman School of Medicine) 홈페이지: http://www.tsirigos.com
Tsirigos Lab의 주요 연구 방향에는 염색질, 후생유전학 및 정밀 의학 응용 분야의 기계 학습이 포함됩니다.
위 내용은 게놈 형태 예측 모델 및 고처리량 컴퓨터 유전자 스크리닝 방법 탐색 및 응용의 상세 내용입니다. 자세한 내용은 PHP 중국어 웹사이트의 기타 관련 기사를 참조하세요!