
Nature sub-journal, AlphaFold보다 우수, 전체 원자 샘플링, 펩타이드 구조 예측을 위한 AI 방법

- WBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWB원래의
- 2024-07-11 11:49:39969검색

딥 러닝 방법은 생체 분자 구조의 단일 상태 예측에 상당한 진전을 가져왔습니다. 그러나 생체분자의 기능성은 그들이 가정할 수 있는 형태의 범위에 따라 달라집니다. 이는 다양한 생물학적 과정에 관여하고 치료제로서 큰 관심을 끄는 매우 유연한 분자 종류인 펩타이드의 경우 특히 그렇습니다.
토론토 대학의 Philip M. Kim과 Osama Abdin은 입력 펩타이드의 허용된 구조 공간에서 직접 모든 원자 샘플링을 가능하게 하는 전송 가능한 생성 모델인 PepFlow를 개발했습니다. 연구원들은 확산 프레임워크에서 모델을 훈련한 다음 구조적 샘플링을 위해 등가 흐름을 사용했습니다.
일반화된 전체 원자 모델링의 엄청난 비용을 극복하기 위해 생성 프로세스를 모듈화하고 슈퍼 네트워크를 통합하여 시퀀스별 네트워크 매개변수를 예측했습니다. PepFlow는 펩타이드 구조를 정확하게 예측하고 기존 방법보다 짧은 실행 시간에 실험적인 펩타이드 컬렉션을 효율적으로 재현합니다. PepFlow는 거대고리화와 같은 제약 조건을 충족하는 형태를 샘플링하는 데에도 사용할 수 있습니다.
"지금까지 우리는 펩타이드의 전체 형태를 시뮬레이션할 수 없었습니다." 연구의 제1저자인 오사마 압딘(Osama Abdin)은 "PepFlow는 딥 러닝을 사용하여 몇 분 안에 펩타이드의 정확한 형태를 포착합니다. 모델은 설계에 의해 약물 개발을 안내하는 바인더로 펩티드를 사용할 수 있는 잠재력을 가지고 있습니다."
이 연구의 제목은 "하이퍼네트워크 조절 확산을 통한 펩티드 에너지 환경에서 직접 구조 샘플링"이며 6월 27일 "Nature Machine Intelligence"에 게재되었습니다. 2024.

- 단백질-펩타이드 상호작용은 분자 경로에 널리 퍼져 있으며 세포 기능에 중요합니다.
- 단백질-단백질 상호작용의 최대 40%는 펩타이드 결합과 관련됩니다.
- 펩타이드는 짧은 서열의 구형 단백질을 무질서한 영역과 결합하여 작동합니다.
펩타이드의 치료 잠재력
- 펩타이드는 특이성이 높고 독성 위험이 낮습니다.
- 펩타이드는 생물학적 제제에 비해 생산 비용이 저렴하고 면역원성이 약합니다.
- 펩타이드 요법은 제약 시장에서 점유율을 계속 확대하고 있습니다.
펩타이드 모델링 및 엔지니어링
- Philip M. Kim은 PepFlow 모델이 펩타이드에 초점을 맞춘다고 말했습니다. 펩타이드는 자연 활동을 하는 중요한 생체 분자이고 기능을 이해하려면 펩타이드의 구조를 시뮬레이션해야 하기 때문입니다.
- Philip M. Kim과 Osama Abdin이 펩타이드 형태의 직접 전체 원자 샘플링 방법을 제안합니다.
- 정확하고 효율적인 전체 원자 샘플링은 짧은 펩타이드의 경우에도 매우 어렵습니다.
그림: PepFlow 아키텍처의 개략도. (출처: 논문)
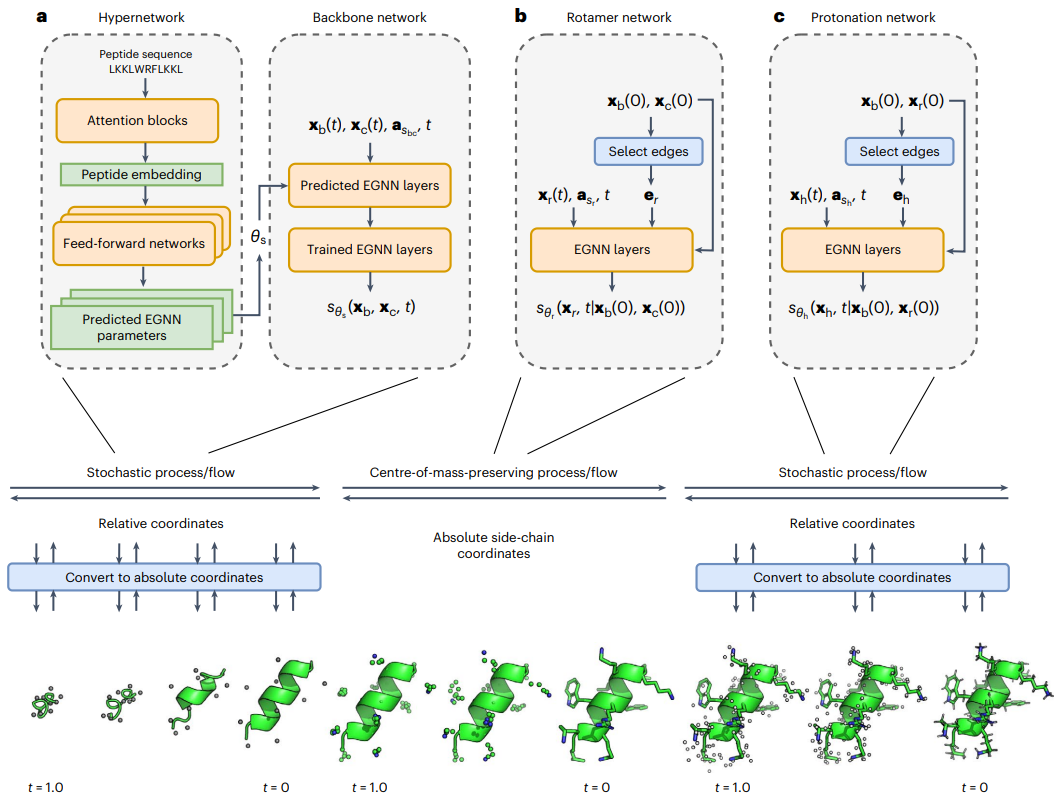
이 문제를 해결하기 위해 그들은 모든 입력 펩타이드 서열에 대한 모든 원자 형태를 예측할 수 있는 모듈식 하이퍼네트워크 조건 생성 모델인 PepFlow를 개발했습니다. PepFlow는 알려진 분자 구조에 대해 학습된 연속 시간 확산 모델입니다. 해당 확률적 흐름 ODE는 에너지 샘플링 및 훈련에 사용됩니다.
PepFlow는 단일항 펩타이드 구조와 짧은 선형 모티프(SLiM) 모음을 예측하는 강력한 기능을 갖추고 있으며, 잠재 공간 구조 검색을 통해 거대고리화와 같은 제약 조건 하에서 펩타이드 구조를 모델링할 수 있습니다.
이 모델은 최고의 Google Deepmind AI 시스템인 AlphaFold의 기능을 확장하여 단백질 구조를 예측합니다. PepFlow는 주어진 펩타이드에 대해 다양한 형태를 생성함으로써 AlphaFold2보다 성능이 뛰어납니다. AlphaFold2는 이 문제를 해결하도록 설계되지 않았습니다.
PepFlow를 차별화하는 것은 그 뒤에 숨은 기술 혁신입니다. 예를 들어, 매우 발전된 물리 기반 기계 학습 모델인 볼츠만 생성기(Boltzmann Generator)에서 영감을 받은 일반화된 모델입니다.
"PepFlow 모델링을 사용하면 펩타이드의 실제 에너지 상태에 대한 통찰력을 얻을 수 있습니다." Abdin은 "PepFlow를 개발하는 데 2년 반이 걸렸고 훈련하는 데는 단 한 달이 걸렸지만 다음 단계로 나아갈 가치가 있습니다. 펩타이드의 한 가지 구조만 예측하는 모델 전반적으로 펩타이드 형태를 정확하고 효율적으로 샘플링하는 능력은 펩타이드 도킹 및 설계를 향상시킬 수 있는 잠재력을 가지고 있습니다. 펩타이드 도킹 방법은 일반적으로 관심 있는 단백질에 도킹된 펩타이드 형태의 라이브러리로 시작됩니다. 보다 정확한 펩타이드 앙상블 생성은 이 프로세스를 개선할 수 있습니다.
PepFlow는 또한 표적 단백질-단백질 경계면에서 형태를 가정하기 위한 다양한 서열의 성향을 평가하는 데 사용될 수 있으며, 이는 차례로 억제 펩타이드를 설계하는 데 사용될 수 있습니다.
그림: PepFlow와 분자 역학 시뮬레이션으로 생성된 앙상블 비교. (출처: 논문) 
PepFlow has a significant drawback, unlike the Boltzmann generator, PepFlow lacks the ability to reweight the generated samples to achieve an accurate Boltzmann distribution.
While PepFlow is capable of performing likelihood calculations on generated samples, tractable calculations require the use of stochastic estimators, which adds noise to the calculated values. Additionally, PepFlow occasionally generates high-energy samples but is unable to capture the full energy landscape observed in molecular dynamics simulations.
One potential way to improve PepFlow is to transfer the developed model to other sampling frameworks. A normalized flow was used in the conditional settings and different sampling methods were used to facilitate sampling from the Boltzmann distribution.
The flow matching paradigm recently developed by the academic community further serves as an alternative to training continuous normalized flow models in a simulation-free manner. Flow matching has been effectively used for structural sampling of different molecules, including small molecules and proteins, and can potentially be used to extend the effectiveness of the PepFlow framework.
In summary, PepFlow is designed to be easily extensible to account for other factors, new information, and potential uses.
Even in just a first version, PepFlow is a comprehensive and effective model with potential for further development of therapeutics that rely on peptide binding to activate or inhibit biological processes.
Paper link: https://www.nature.com/articles/s42256-024-00860-4
Related reports: https://phys.org/news/2024-06-deep-outperforms-google-ai -peptide.html
위 내용은 Nature sub-journal, AlphaFold보다 우수, 전체 원자 샘플링, 펩타이드 구조 예측을 위한 AI 방법의 상세 내용입니다. 자세한 내용은 PHP 중국어 웹사이트의 기타 관련 기사를 참조하세요!