
ホームページ >テクノロジー周辺機器 >AI >幾何学的な深層学習手法を使用して薬物分子を合成するための最適なオプションを予測し、新薬発見への道を開く
幾何学的な深層学習手法を使用して薬物分子を合成するための最適なオプションを予測し、新薬発見への道を開く

- WBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWB転載
- 2024-01-13 22:36:071130ブラウズ

後段階官能化は、薬剤候補の特性を最適化するための経済的な方法です。しかし、薬物分子の化学的複雑さにより、後期段階の機能化が困難になることがよくあります。
この問題を解決するために、ミュンヘン大学、チューリッヒ工科大学、ロシュ バーゼルの研究者が協力して、後期段階の機能化プラットフォームを開発しました。幾何学的深層学習とハイスループット反応スクリーニング技術に基づいています
ホウ素化が機能化の重要なステップの 1 つであることを考慮して、計算モデルを使用してさまざまな反応条件下での収率を予測しました。平均絶対誤差範囲は次のとおりです。 4~5%。このモデルは、既知の基質と未知の基質に対する新しい反応を、それぞれ 92% と 67% の精度で分類できました。分類器の F スコアは 67% で、主生成物の位置選択性を正確に捕捉することができました。私たちは、23 種類の市販薬分子に適用した場合に、構造多様化の多くの機会を特定することに成功しました。
この研究のタイトルは「幾何学深層学習を使用して、後期段階の薬物多様化を促進するハイスループット実験を可能にする」であり、ジャーナルに掲載されました。 Nature Chemistry 2023 年 11 月 23 日

#LSF プロジェクトは医薬化学研究において重要な役割を果たします
構造の新規性と複雑性医薬化学における構造活性相関の確立を目指す場合、化学標的構造の合成は困難になります。構造活性相関モデルは、医薬品候補の薬理学的活性と物理化学的特性を改善するためのリード化合物とリード化合物の最適化計画を導くことができます。効率的な統合は、設計、製造、テスト、分析サイクルのボトルネックである構造活性相関の探索にとって極めて重要です。C-H 結合を活性化および修飾して有機足場を実現するための代替方法は数多くあります。後期段階機能化(LSF):分子ビルディングブロックから先進的な医薬品分子まで。多くの触媒システムは、指向性および無指向性のアプローチに加え、修飾類似体への化学的および部位選択的なアクセスを提供します。数多くの LSF 法の中で、C-H ボリル化法は、高速化合物に最も一般的に使用されると考えられています。アプローチ。有機ホウ素化合物は、その後の C-C 結合カップリング反応の信頼できる手段としてさまざまな官能基に変換できるため、広範な構造活性相関研究が可能になりますしかし、現在、創薬においては、LSF のアプリには報告が少ない。これらのレポートのほとんどは、単一の LSF 反応タイプに焦点を当てています。異なる結合強度、電子的特性、立体および官能基環境を備えた複数の種類の C-H 結合の直接 LSF には課題が生じます。さらに、LSF プロジェクトの実施は時間とリソースを大量に消費することが多く、多くの医薬化学プロジェクトのタイトなスケジュールと限られた資産とは一致しません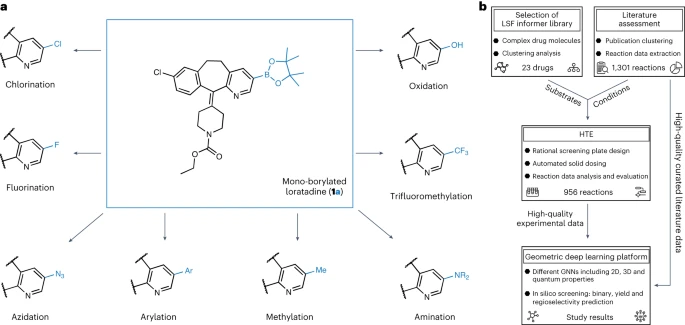
人工知能対応LSF(言語サポート機能)
ハイスループット実験(HTE)は確立された反応最適化手法です。 、小型化された低バッチスクリーニングが可能であり、少数の貴重な構成要素と消耗品を使用して複数の変換を迅速かつ再現性よく並行して実行できます。 HTE は、応答の成功と失敗に関する高品質のデータセットを生成する FAIR (検索可能性、アクセシビリティ、相互運用性、再利用性) ドキュメントと組み合わせることで、高度なデータ分析と機械学習を可能にし、創薬のための LSF を可能にします。 グラフ ニューラル ネットワーク (GNN) は、分子の特徴抽出と属性予測に広く使用されています。化学反応計画のために開発されたさまざまな機械学習手法の中で、GNN は逆合成計画、位置選択性予測、反応生成物の予測に適用されて成功しています。さらに、同様の問題を解決するために、トランスフォーマーやフィンガープリントに基づく方法も開発されています。研究によると、遷移状態の幾何学構造を学習することで、競合する反応の結果を正確に予測できることが示されています。電子効果によって駆動される反応の位置選択性の予測は、密度汎関数理論 (DFT) と原子の部分電荷のグラフによる特性評価を使用して改善できます。グラフ機械学習とハイスループット実験 (HTE) を組み合わせることで、有機基板の C-H 活性化反応の条件を最適化できます。一部の研究では、場合によってはエナンチオ選択性を含む反応結果を予測する能力を持つ遷移状態の深層学習モデルの使用に焦点を当てていますが、これらの方法は小分子構造および比較的小さな分子に限定されています。このようなモデルをより構造的に複雑な薬物様分子に適用することは困難です。文献研究に基づいて、イリジウム触媒によるホウ素化反応の位置選択性は、遷移状態の量子化学情報で強化されたハイブリッド機械学習モデルを通じて予測できます。ただし、C-H 活性化反応モデルのパフォーマンスと複数の芳香環系を持つ分子における位置選択的応用に対する立体効果と電子効果の影響はまだ調査されていません。幾何学深層学習による自動 LSF ホウ素スクリーニング
ミュンヘン大学、チューリッヒ工科大学、ロシュ ファーマシューティカルズ バーゼルの研究者は、幾何学深層学習 LSF ホウ素に適用された自動 LSF ホウ素スクリーニング手法を発表します。後期段階のヒット商品とリード多様化の機会を特定するためのスクリーニング方法。コンピューターによる深層学習を利用して、複雑な薬物分子 LSF の反応結果、収率、位置選択性を予測しました。
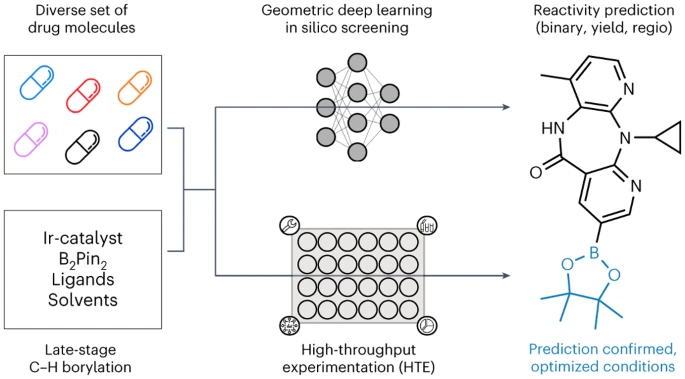
以上が幾何学的な深層学習手法を使用して薬物分子を合成するための最適なオプションを予測し、新薬発見への道を開くの詳細内容です。詳細については、PHP 中国語 Web サイトの他の関連記事を参照してください。