
ホームページ >テクノロジー周辺機器 >AI >ウィスコンシン大学と中国のチームの新しいマルチモーダル データ分析および生成方法 JAMIE は、細胞の種類と機能の予測能力を大幅に向上させます
ウィスコンシン大学と中国のチームの新しいマルチモーダル データ分析および生成方法 JAMIE は、細胞の種類と機能の予測能力を大幅に向上させます

- WBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWB転載
- 2023-06-10 14:43:291487ブラウズ
近年、単一細胞技術の急速な発展により、単一細胞のさまざまな特性を測定して、単一細胞のマルチモーダルデータ(scRNA-seq、scATAC-seq、Patchなど)を取得できるようになりました。 -seq) 。
これらのデータは、細胞機能と分子機構についての洞察を得るのに役立ちます。たとえば、研究者たちは最近、機械学習手法を使用して単一細胞のマルチモーダルデータ間の関係を分析し、細胞の種類や疾患に関与する生物学的メカニズムを理解しています。
ただし、単一セルのマルチモーダル データの取得にはコストがかかることが多く、モーダル損失が頻繁に発生します。既存の機械学習手法は通常、データの充填と埋め込みに完全に一致したマルチモーダル データを必要とし、モダリティが欠落している状況には適していません。
この問題を解決するために、ウィスコンシン大学マディソン校の Wang Daifeng の研究室は、ジョイント変分オートエンコーダに基づくオープンソースの機械学習手法を開発しました - マルチモーダル代入および埋め込み用ジョイント変分オートエンコーダ(ジェイミー)。
JAMIE は、データのアライメント、埋め込み、欠損データの補完など、単一細胞のマルチモーダル データの統合分析に使用して、細胞の種類と機能をより適切に予測できます。
この研究は、最近 Nature Machine Intelligence に掲載されました。

論文アドレス: https://www.nature.com/articles/s42256-023-00663 -z
プロジェクトアドレス: https://github.com/daifengwanglab/JAMIE
JAMIE メソッドの紹介
JAMIE は、再利用可能な関節変分オートエンコーダ モデルをトレーニングし、利用可能なマルチモーダル データを同様の潜在空間能力に個別に投影することで、シングルモーダル パターン推論を強化します。
図 1 に示すように、クロスモーダル代入を実行するために、JAMIE はデータをエンコーダーに供給し、反対側のデコーダーを通じて潜在空間の結果を処理します。
JAMIE は、再利用可能で柔軟なオートエンコーダーの潜在空間生成とアライメント手法の自動対応推定を組み合わせて、不完全な対応を持つマルチモーダル データの処理を可能にします。
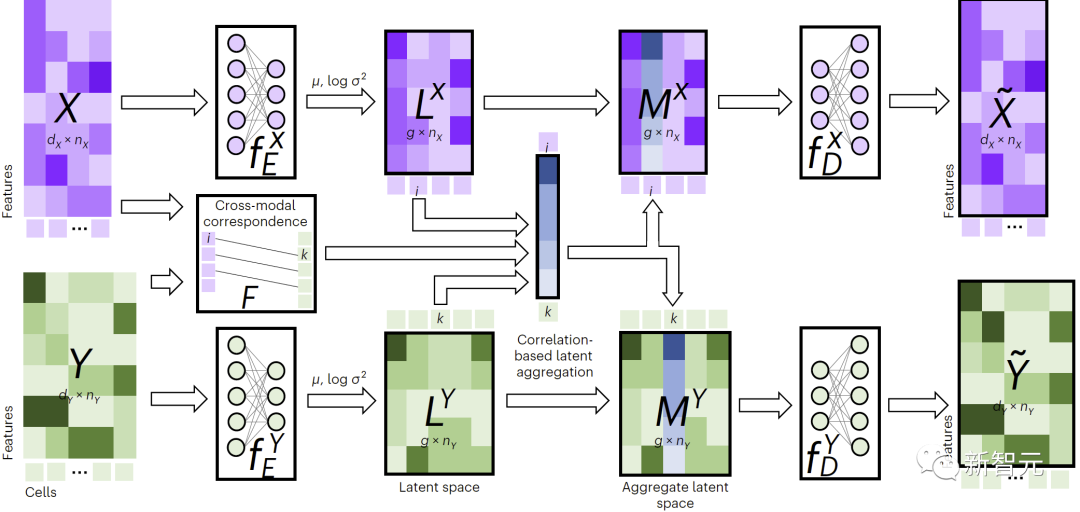
#図 1. JAMIE メソッドの概要
具体的には、JAMIE は次の 2 つのステップに分割できます:
- 入力データの前処理。二峰性モードを例として、各モードに対応するデータ行列がそれぞれ と であると仮定します。ここで、特徴の次元と合計は異なる可能性があり、サンプルの数も異なる可能性があることに注意してください。前処理では、各行列の各行が平均 0、分散 1 になるように正規化されます。対応するデータがある場合、ユーザーはモーダル相関行列を提供してパフォーマンスを向上させることができます。ここで、モーダル内の 番目のサンプルがモーダル内の 番目のサンプルに完全に対応することを意味し、既知の対応関係がないことを意味し、部分的な対応関係があることを意味します。対応。 。
- 関節変分オートエンコーダーを使用して、各モダリティの類似性潜在空間を学習します: と 、ここで (デフォルト、ユーザー調整可能) は潜在空間次元です。トレーニング プロセス中に、JAMIE は次の損失関数を最小化します。
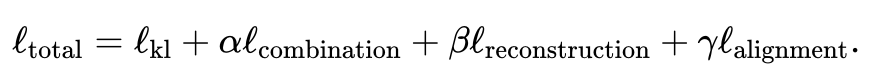
合計損失関数には 4 つの項目が含まれます。
最初の項目は、変分オートエンコーダーによって推定された分布と多変量標準正規分布の間のカルバック・ライブラー (KL) 発散を計算します。これは、潜在空間と空間の連続性を保存するのに役立ちます。 。
各項目の具体的な表現については論文原文を参照してください。最初の項目に対する 2 番目、3 番目、および 4 番目の項目の重みはユーザーが調整でき、JAMIE は一般的な状況に適したデフォルトの重みも提供します。
次の表は、JAMIE のモデルと適用範囲と現在の最先端の手法との比較を示しています。 JAMIE は、複数の異なる統合および補間方法の機能を単一のアーキテクチャに統合することで、欠落モダリティの補間を可能にし、その結果、非オミクス データの互換性が得られ、部分的な対応のみでマルチモーダル データを処理できるようになります。
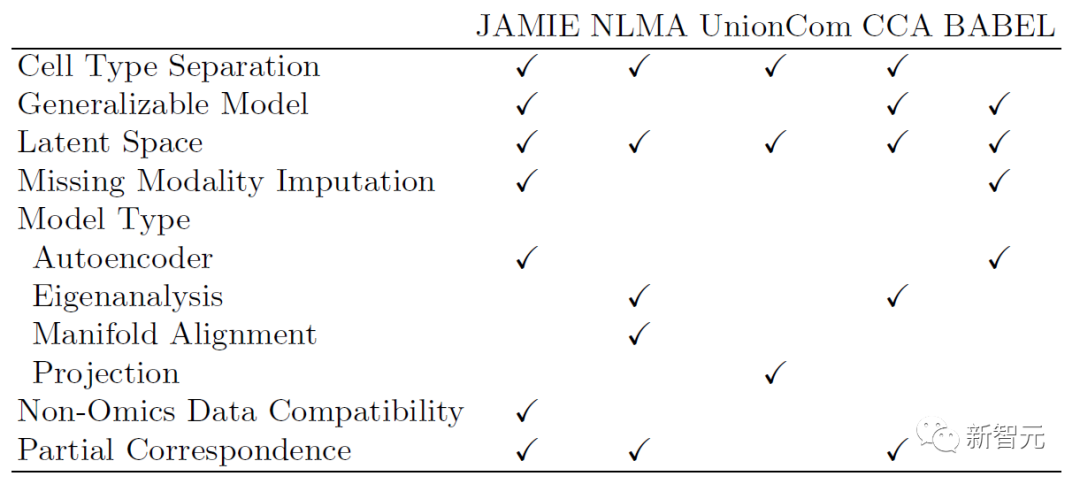
#表 1. さまざまなマルチモーダル統合と欠落モーダル充填方法の比較。 JAMIE は、単一のアーキテクチャを通じて、複数の異なる統合および補間方法の機能を統合します。 NLMA: 非線形多様体アライメント [15]、UnionCom [7]、CCA: 正準相関分析 [15、16]、BABEL [5]。
JAMIE の主な用途マルチモーダル データ統合と表現型予測
統合多峰性データの活用により、分類パフォーマンスが向上し、表現型の知識と複雑な生物学的メカニズムの理解が強化されます。
2 つのデータ セット と対応する関係が与えられると、JAMIE はトレーニングされたエンコーダー と に基づいて潜在空間データ を生成し、 に基づいてクラスタリングまたは分類を実行できます。
潜在空間データに基づくクラスタリングには、両方のモダリティを特徴生成に組み込むなど、いくつかの利点があります。 JAMIE は、細胞型の予測など、サンプルの対応関係を予測できます。
部分的にラベル付けされたデータ セットの場合、同じクラスター内のセルは同様のタイプを持つ必要があります。
JAMIE は、潜在空間データを生成するプロセスでさまざまなタイプのデータの特性を分離するため、通常、より良い結果を得るために複雑なクラスタリングや分類アルゴリズムは必要ありません。
高次元データの場合、JAMIE は細胞型クラスタリングの視覚化に UMAP [32] を使用します。
クロスモーダル データ入力
現在、クロスモーダル データ入力の多くの方法では、彼らが水増しの目的で基礎となる生物学的メカニズムを学習したことを実証します。
フィードフォワード ネットワークや線形回帰手法と比較して、JAMIE は基礎となる生物学的メカニズムをよりよく学習し、より厳密な数学的基礎に基づいて欠損データを予測できます。
図 2 は、JAMIE のクロスモーダル データ入力プロセスを示しています。 JAMIE は、まずトレーニング データでエンコード モデルとデコード モデルをトレーニングします。
新しいデータの場合、JAMIE はまずデータから学習したエンコーダーを使用して潜在空間に投影して を取得し、次に潜在空間特徴を集約して取得し、最後に対応するデコーダーを使用してデータが欠落パターンにデコードされました。
JAMIE は潜在空間を使用して細胞間の対応を予測します。これは、データの特徴と表現型の関係を理解するのに役立つ可能性があります。
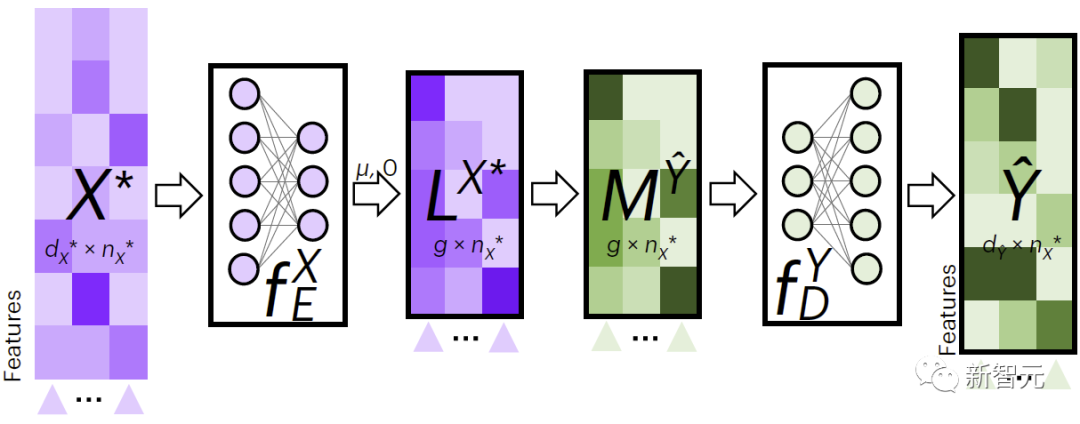
#図 2. JAMIE クロスモーダル補間
潜在空間特徴と充填特徴の説明
訓練されたモデルを説明するために、JAMIE は SHAP (SHapley Additive exPlanations) を使用しました。 )[18]。SHAP は、モデルによって生成された個々の予測をサンプル変調することによって、個々の入力特徴の重要性を評価します。これは、さまざまな興味深いアプリケーションに使用できます。
ターゲット変数を表現型によって簡単に分離できる場合、SHAP はさらなる研究に関連する特性を特定できます。さらに、代入を実行すると、SHAP はモデルによって学習されたクロスモーダル接続を明らかにできます。 モデルとサンプルが与えられた場合、背景特徴ベクトルがどこにあるかなどの SHAP 値を学習します。 の場合、SHAP 値とバックグラウンド出力の合計は に等しくなります。ここで、それぞれはモデル出力への影響に比例します。 もう 1 つの有用な手法は、分類 (例: LTA [7,19]) または代入 (例: 代入された特徴と測定された特徴の関係の間の対応) のための重要なメトリックを選択し、モデル内の各特徴を 1 つずつ削除 (バックグラウンド値に置き換え) することで、メトリクスを取得します。 そして、主要な指標が悪化した場合、これは、削除された特徴がモデルの結果にとってより重要であることを示します。 JAMIE は、検証のために一般的に使用される 4 つの単一細胞マルチモーダル データ セットを使用しました。 (1) MMD-MA からの分岐多様体のガウス分布サンプリングによって生成されたシミュレートされたマルチモーダル データ (300 サンプル、3 つのセル タイプ); (2) マウス視覚野 (3,654 サンプル、6 細胞タイプ) およびマウス運動野 (1,208 サンプル、9 細胞タイプ) の単一神経細胞からのパッチシーケンス遺伝子発現および電気生理学的特徴データ; (3) ヒトの発達中の脳からの 8,981 個のサンプル (在胎 21 週、ヒトの大脳皮質をカバーする 7 つの主要な細胞タイプ) 10x 単一細胞マルチオミクス遺伝子発現およびクロマチン アクセシビリティ データ; (4) COLO-320DM 結腸腺癌細胞株の 4,301 細胞からの scRNA-seq 遺伝子の発現および scATAC-seq クロマチン アクセシビリティ データ。 評価の結果、JAMIE が他の方法よりも大幅に優れていることがわかりました (図 3 の MMD-MA の分岐多様体シミュレーション データの結果の比較、および図 4 のマウス視覚野データの結果の比較) )およびマルチモーダル充填の重要な機能が優先され、細胞解像度レベルで潜在的に新しいメカニズムの洞察が提供されます。 実験結果
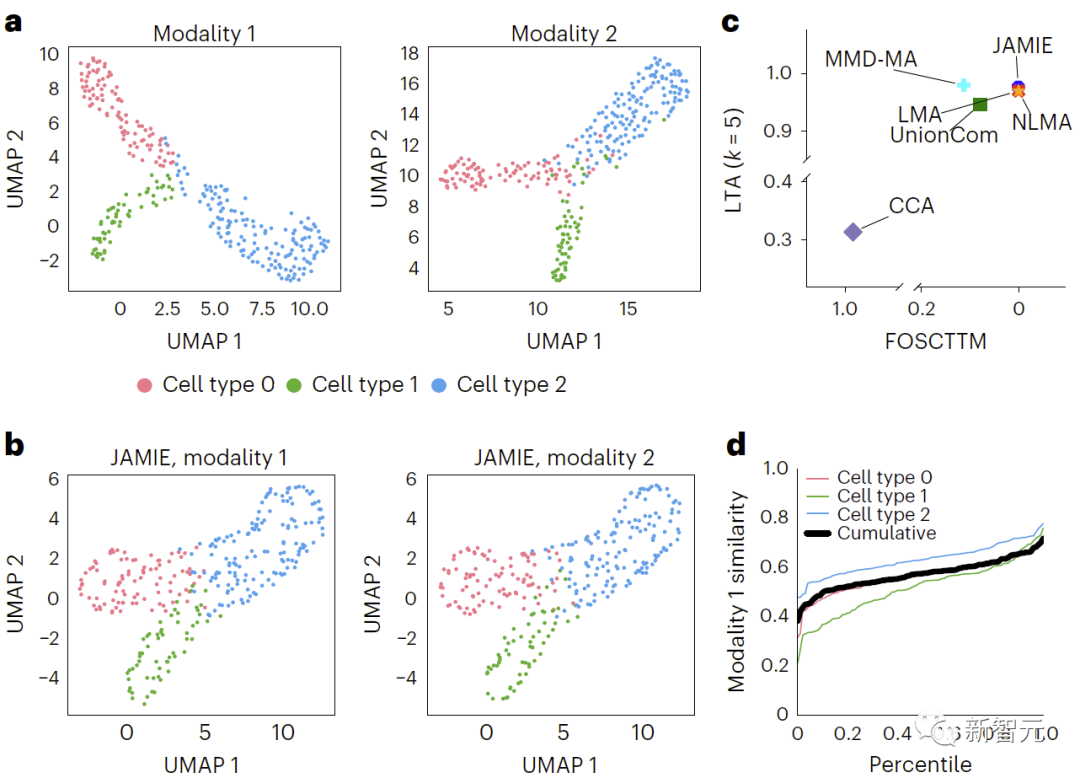
#図 3 では、生の空間データに UMAP アルゴリズムを適用し、さまざまなセルに応じて色付けしています。タイプ 、シミュレートされたマルチモーダル データの結果を比較しました。 b. JAMIE 潜在空間の UMAP。 c. セルタイプ分離に利用可能なすべての対応情報を使用する場合の JAMIE と既存の技術 (CCA[15,16]、LMA[15]、MMD-MA[8]、NLMA[15]、および UnionCom[7])) を比較します。 X 軸は真の平均に近いサンプルの割合、Y 軸は LTA[7,19] 値です。モード 1 では、1-JS 距離の累積分布を計算して、測定値と補間値の類似性を評価します。色付きの各線は特定の細胞タイプの類似性を表し、黒い線は細胞タイプ間の平均類似性を表します。
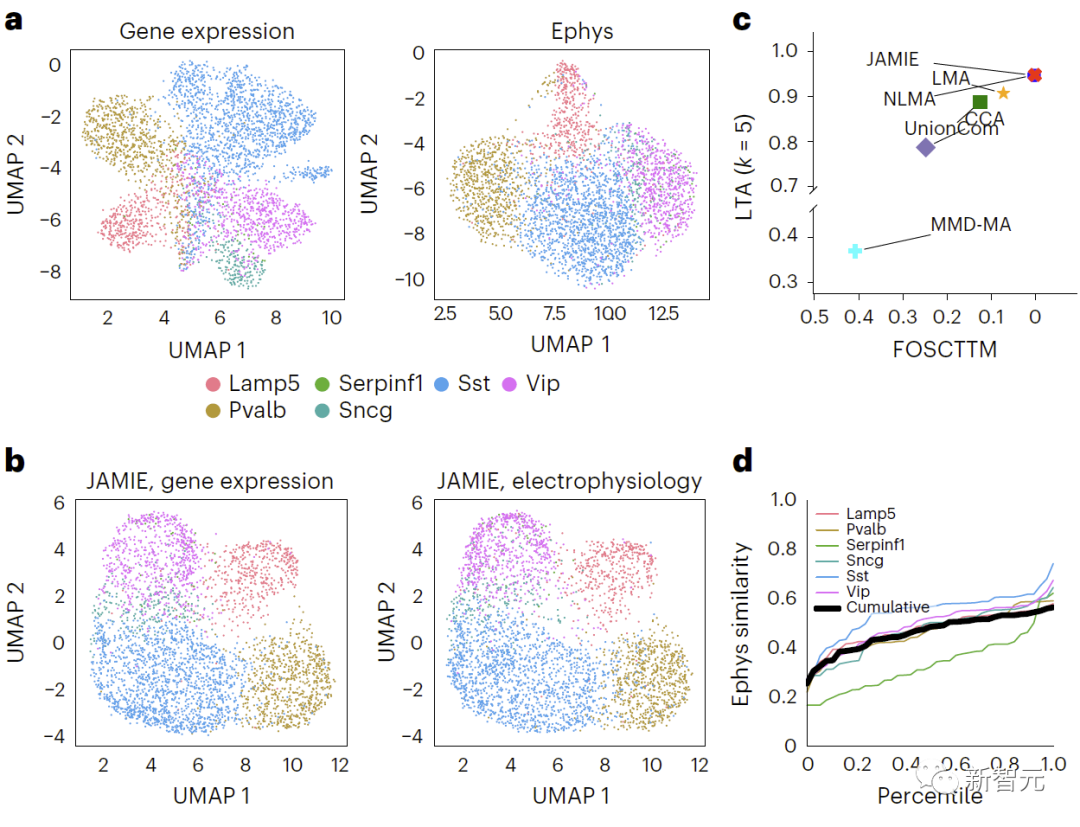
再掲: マウス視覚野における遺伝子発現と電気生理学的特性の結果の比較、異なるセルタイプは、元の空間で UMAP を使用して色付けされます。図 4 に比較結果を示します。 b. JAMIE 潜在空間の UMAP。 c. セルタイプ分離に利用可能なすべての対応情報を使用する場合の JAMIE と既存の技術 (CCA[15,16]、LMA[15]、MMD-MA[8]、NLMA[15]、および UnionCom[7])) を比較します。 X 軸は真の平均に近いサンプルの割合、Y 軸は LTA[7,19] 値です。モード 1 では、1-JS 距離から計算された測定値と補間値の間の類似度の累積分布が調査されます。色付きの各線は 1 つの細胞タイプの類似性を表し、黒い線は異なる細胞タイプの平均類似性を表します。
概要要約すると、JAMIE は、単一セルのマルチモーダル データを統合予測するための新しいディープ ニューラル ネットワーク モデルです。
ジョイント変分オートエンコーダ (VAE) 構造に依存する新しい潜在埋め込み集約手法を通じて、複雑な、混合された、または部分的に対応するマルチモーダル データに適しています。前述の優れたパフォーマンスに加えて、JAMIE は効率的なコンピューティング機能と低いメモリ使用量要件も備えています。さらに、事前トレーニングされたモデルと学習されたクロスモーダル潜在埋め込みは、下流の分析で再利用できます。
もちろん、大規模なデータセットの場合、変分オートエンコーダー (VAE) のトレーニングには多くの時間がかかります。したがって、JAMIE の自動 PCA などの以前の特徴選択方法は、時間要件を軽減するのに役立ちます。 VAE では再構成損失が使用されるため、大規模なフィーチャまたは繰り返しフィーチャが低次元の埋め込みフィーチャに不均衡な影響を与えることを避けるために、データの前処理も重要です。特定のクロスモーダル代入の場合、最終モデルにバイアスがかかり、一般化能力に悪影響が及ばないよう、トレーニング データセットの多様性を慎重に考慮する必要があります。 JAMIE は、異なる条件下での遺伝子発現データなど、異なるモダリティではなく異なるソースからのデータセットを調整するように拡張できる可能性もあります。
著者紹介
この論文の著者は、Noah Cohen Kalafut (コンピュータ サイエンス学科博士課程学生)、Huang Xiang (上級研究員)、およびWang Daifeng (PI) は、ウィスコンシン大学マディソン校生物統計学および医療情報学部、コンピューターサイエンス学部、およびワイズマン研究センターに所属しています。責任著者は王大峰教授です。
1973 年に設立されたワイズマン センターは、半世紀にわたって人間の発達、神経発達障害、神経変性疾患の研究を進めてきました。
以上がウィスコンシン大学と中国のチームの新しいマルチモーダル データ分析および生成方法 JAMIE は、細胞の種類と機能の予測能力を大幅に向上させますの詳細内容です。詳細については、PHP 中国語 Web サイトの他の関連記事を参照してください。