
ホームページ >テクノロジー周辺機器 >AI >ゲノム構造予測モデルとハイスループットの計算による遺伝子スクリーニング法の探索と応用
ゲノム構造予測モデルとハイスループットの計算による遺伝子スクリーニング法の探索と応用

- PHPz転載
- 2023-05-08 14:16:08829ブラウズ

#図 0
#さまざまな種類の細胞におけるゲノム構造の違いが遺伝子の特異性を決定します式 、それによって異なる細胞型の機能の違いを決定します。長い間、in situ ハイブリダイゼーションから Hi-C や micro-C テクノロジーなどのハイスループット検出に至るまで、ゲノム構造検出の実験方法は、通常、時間と労力がかかり、コストがかかり、大きな技術的制限がありました。これらの方法は、ゲノム立体構造研究の分野、特に希少な細胞タイプの研究や、大規模なゲノム立体構造制御の因果関係を検証する必要性におけるこれらの実験技術の幅広い応用を大きく制限します。これらの方法の限界により、三次元ゲノム構造制御の分野における新しい発見も長い間制限されてきました。

写真 1##2023 年 1 月 9 日、
NYU グロスマン スクール医学博士のアリストテリス・ツィリゴス研究室とマサチューセッツ工科大学ブロード研究所およびハーバード大学のシア・ボー研究室が協力して、Nature Biotechnology 誌に論文を発表しました。「3D クロマチン組織の細胞型特異的予測により、ハイスループットのインシリコ遺伝子スクリーニングが可能になります。」
 論文アドレス: https://www.nature.com/articles/s41587-022-01612-8
論文アドレス: https://www.nature.com/articles/s41587-022-01612-8
この研究では、筆頭著者であるニューヨーク大学医学部博士課程の学生である Tan Jimin 氏と Xia Bo 博士が、新しいマルチモーダル機械学習モデル C.Origami を初めて提案しました。特定の細胞型のクロマチン構造を解明し、細胞型固有の機能を同定するための遺伝子スクリーニングの原理に基づいた、新しいハイスループット計算遺伝スクリーニング (ISGS) 法を提案します
ゲノム要素は、クロマチン構造の新しいメカニズムの発見に役立ちます規制。#図 2
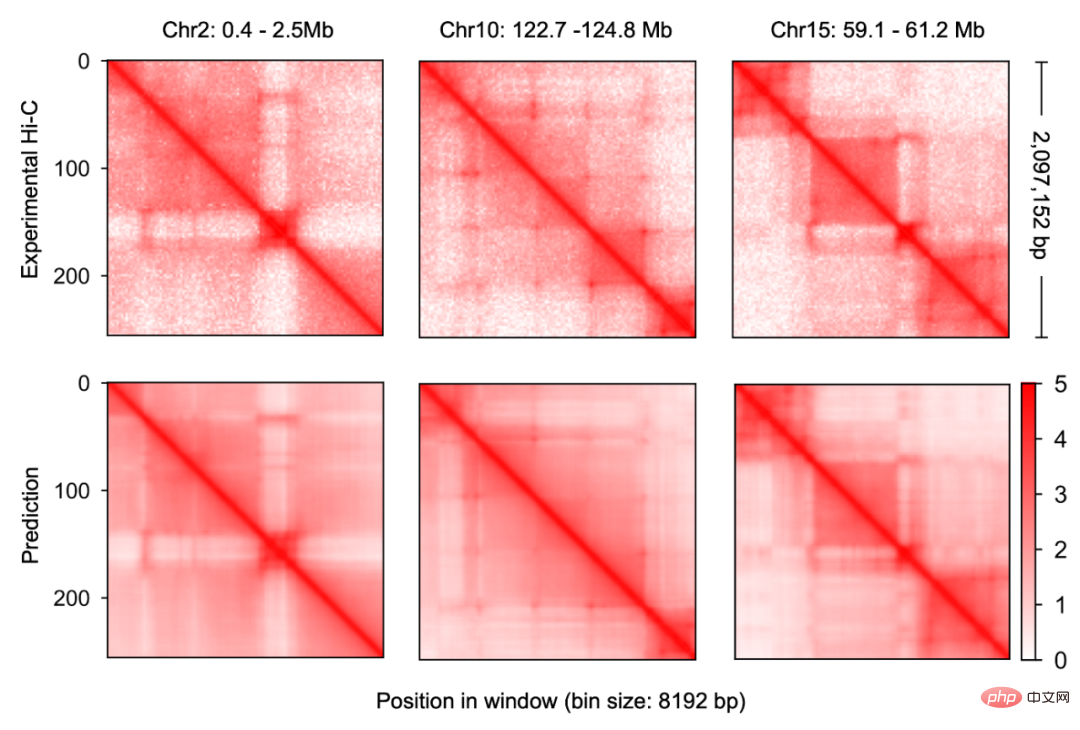
研究者最初の A新しいマルチモーダル深層学習フレームワーク、Origami がゲノム データ用に構築され、DNA 配列情報と細胞固有の機能ゲノム情報を効果的に統合して、新しいゲノム情報を予測できるようになりました。研究者らは、デバッグとモデルトレーニングを繰り返すことで、DNA配列、CTCF結合状態(CTCF ChIP-seq)、ATAC-seqシグナルを入力情報として統合し、二次元Hi-Cマトリックスをクロマチン構造を正確に予測できることを発見した。出力対象を予測します(図1-2)。入力情報は 200 万塩基対の DNA、CTCF ChIP-seq、ATAC-seq でした。研究者は Onehot エンコーディングを使用して離散 DNA 配列をエンコードし、CTCF ChIP-seq および ATAC-seq は非離散特徴をエンコードします。
C.Origami モデルは、DNA とゲノム情報を処理および圧縮するエンコーダー、Transformer 中間層、出力 Hi-C デコーダーの 3 つの部分に分かれています。 #####。エンコーダーは、200 万塩基対の入力情報をエンコードして圧縮するための一連の 1D ResNet とストライド畳み込みで構成されます。エンコーダの最後で、200 万長のメッセージが 256 長に圧縮され、Transformer への入力メッセージとして使用されます。 Transformer のセルフアテンション メカニズムは、異なるゲノム領域間の相互依存性を処理し、モデルの全体的なパフォーマンスを向上させることができます。 Transformer のアテンション マトリックスによって、モデルの解釈可能性も向上します。研究者らは、予測時にモデルがさまざまな領域に重点を置くかを測定するために、注意の重みを「注意スコア」に変換しました。最後に、研究者らは、「外部連結」を使用して、Transformer モジュールの 1D 出力を 2D 接触/隣接行列に変換し、これを Hi-C デコーダの入力情報として使用しました。デコーダは Dirated 2D ResNet です。研究者らは、最終層の各ピクセル位置の受容野がすべての入力情報をカバーできるように、さまざまな層の拡張係数を調整しました。 クロマチン構造を予測するためのこのモデルは、C.Origami と呼ばれます。研究者らは、C.Origami をゲノミクス初のマルチモーダル深層学習モデルと呼んでいます。 C.Origami はそのマルチモーダルな性質により、これまで暴露されたことのない新しい種類の細胞のクロマチン構造を正確に予測 (新規予測) することができます。たとえば、IMR-90 細胞 (肺線維芽細胞) でトレーニングされたモデルは、GM12878 細胞 (B リンパ球) の特定のクロマチン立体構造を正確に予測できました (図 3)。
#図 3 構造変異体 --- - 染色体など転座 - 腫瘍では非常に一般的で、多くの場合クロマチン相互作用パターンを変化させ、がん遺伝子や腫瘍抑制遺伝子の発現に影響を与える可能性があります。クロマチンの立体構造や遺伝子発現に対するこれらの構造変化の影響を研究することは、腫瘍の発生と進行のメカニズムを理解するために重要です。この種の研究では通常、構造変異部位のクロマチン立体構造を分析するために 4C-seq や Hi-C などの実験を使用する必要がありますが、多くの場合、リソースと時間に制限があり、大規模に実施することが困難です。 この研究では、C. Origami は入力変数の DNA 配列の変動をモデル化し、変異したがんゲノムにおける新しいクロマチン相互作用を予測できます。以前の研究では、T 細胞急性リンパ性白血病 (T-ALL) 細胞モデル CUTLL1 には chr7-chr9 染色体転座があることが判明しました (図 4)。 C. Origami は、染色体転座変異体をコンピューターでシミュレーションすることにより、変異部位の新しい TAD 構造を正確に予測し、chr9 から chr7 に伸びる「クロマチン ストライプ」構造を検出しました (図 4)。 #図 4 C の正確な予測効果を考慮して.Origami, 逆遺伝学的スクリーニングの原理に触発されて、研究者らは細胞型特異的な機能的ゲノム要素を体系的に特定し、新しいものの発見に役立つ新しいハイスループット計算遺伝的スクリーニング (ISGS) 方法を提案しました。 調節分子の染色 (図 5)。研究者らは、クロマチンの立体構造に必要なシス調節要素を体系的に同定するための、C. Origami モデルに基づく計算遺伝スクリーニング ISGS のフレームワークを開発しました。著者らは、ゲノムワイド 1kb 分解能の ISGS を使用して、クロマチンの立体構造に重要な影響を与えるシス制御要素 (ゲノムの約 1%) を単離しました。これらのクロマチン構造調節配列は、CTCF結合およびATAC-seqシグナルに対して異なる依存性を示します(図5)。 #図 5 ISGS フレームワークにより、細胞または疾患に特異的なクロマチン立体構造のハイスループット スクリーニングが可能になります。研究者らは、CUTLL1、Jurkat、および正常な T 細胞で ISGS を実行し、CHD4 遺伝子近くのシス調節要素 (CHD4-insu) が T-ALL 細胞で特異的に失われていることを発見しました。スクリーニング結果は、T-ALL 細胞における CHD4-insu の絶縁喪失により、CHD4 遺伝子が新たなクロマチン相互作用を確立できるようになり、それによって CHD4 発現が上方制御され、白血病細胞の増殖が促進される可能性があることを示しています。 ISGS は、クロマチンの立体構造を制御する新規トランス作用因子を系統的に発見するためにも使用できます。研究者らは、重要な細胞型特異的な制御配列と転写因子結合部位の濃縮分析を通じて、細胞型特異的なゲノム構造に寄与する制御因子を同定した。興味深いことに、以前の研究では、MAZがCTCFと一緒にクロマチンの立体構造を調節する可能性があることが判明しました。著者らは、ISGSおよび転写因子濃縮分析を通じて、MAZは開いたクロマチン領域では非常に濃縮されているが、CTCFが結合する非開いたクロマチン領域では弱い結合しか示さないことを発見した。この結果は、MAZ が CTCF とは独立してゲノム構造を調節している可能性があることを示唆しています。 研究者らは、クロマチン構造予測において DNA 配列とクロマチン情報を組み合わせるマルチモーダル機械学習モデルに大きな可能性があると考えています。モデル Origami の基礎となるマルチモーダル アーキテクチャは、エピジェネティック修飾、遺伝子発現、突然変異の機能スクリーニングなど、他のゲノミクス データのアプリケーションに拡張できます。研究者らは、将来のゲノミクス研究は、生物学的実験によって検証された新世代のハイスループット研究手法によって補完された、一次計算遺伝子スクリーニングのツールとしてディープラーニングモデルの使用にさらに移行すると予測しています。 この研究では、ニューヨーク大学医学部の博士候補者である Tan Jimin が筆頭著者であり、Aristotelis Tsirigos 博士と Xia Bo 博士が共同責任者です。著者たち。この研究は、2020年10月の疫病封鎖中のXia BoとTan Jiminのブレインストーミングから始まりました。2年半の改善と磨きを経て、2023年1月にNature Biotechnologyに正式に掲載されました。 このプロジェクトのコードとトレーニング データは GitHub と Zenodo でオープンソース化されており、機能デモンストレーション用に Google Colab が装備されています。 #プロジェクトアドレス: https://github.com/tanjimin/C.Origami Xia Bo 博士の研究室 (MIT とハーバード大学のブロード研究所) ホームページ: www.boxialab.org Xia Bo 博士は、中核となるメカニズムと制御の分析に取り組んでいます。ゲノムの三次元構造、ヒトの病気、発生、進化に対する生物学的重要性。 Xia Bo の研究室では、志を同じくする博士研究員のチームへの参加を歓迎しています。 Tsirigos Lab (ニューヨーク大学グロスマン医科大学院) ホームページ: http://www.tsirigos.com Tsirigos Lab のメインページ 研究対象には、精密医療におけるクロマチン、エピジェネティクス、機械学習の応用が含まれます。 


以上がゲノム構造予測モデルとハイスループットの計算による遺伝子スクリーニング法の探索と応用の詳細内容です。詳細については、PHP 中国語 Web サイトの他の関連記事を参照してください。