
ホームページ >テクノロジー周辺機器 >AI >タンパク質機能予測のための新しい SOTA、オックスフォードの上海理工大学などによる統計ベースの AI 手法、Nature サブジャーナルに掲載
タンパク質機能予測のための新しい SOTA、オックスフォードの上海理工大学などによる統計ベースの AI 手法、Nature サブジャーナルに掲載

- PHPzオリジナル
- 2024-08-22 16:45:02826ブラウズ

タンパク質は他の分子と結合して、ほぼすべての基本的な生物学的活動を促進します。したがって、タンパク質の機能を理解することは、健康、病気、進化、生物の機能を分子レベルで理解するために重要です。
しかし、2 億以上のタンパク質はまだ特性が解明されておらず、さまざまな品質のアノテーションを予測するために、計算手法はタンパク質の構造情報に大きく依存しています。
最近、オックスフォード大学、チューリッヒ工科大学、上海科学技術大学、北京師範大学の研究チームは、タンパク質の機能アノテーションと機能部位の同定を促進するために、PhiGnetと呼ばれる統計ベースのグラフネットワーク手法を設計しました。
PhiGnet はパフォーマンスにおいて他のメソッドよりも優れているだけでなく、構造情報がない場合でもシーケンスと関数のギャップを埋めます。この発見は、深層学習を進化データに適用すると、残基レベルで機能部位を強調表示でき、生物医学におけるタンパク質の既存の特性と新しい機能の解釈と研究に貴重なサポートを提供できることを示しています。
関連する研究は「統計情報に基づいたグラフネットワークを使用したタンパク質機能の正確な予測」と題され、8月4日付けの「Nature Communications」に掲載されました。

タンパク質の機能を理解することは、多くの主要な生物学的活動の複雑なメカニズムを理解するために不可欠であり、医学、バイオテクノロジー、医薬品開発分野には広範囲にわたる影響があります。
現在までに、3 億 5,600 万を超えるタンパク質が UniProt データベースで配列決定されています (2023 年 6 月) が、その大部分 (~80%) には既知の機能注釈がありません。
ディープラーニング手法は、タンパク質の 3D 構造を予測する際に驚くべき精度を達成し、ab initio 手法や相同性モデリングなどの古典的な手法の能力を上回ります。しかし、タンパク質に機能的アノテーションを正確に割り当てることは、特に実験的アッセイと比較すると依然として困難です。
これらの課題に対処するために、研究者らは、共進化する残基に含まれる情報を使用して残基レベルの関数に注釈を付けることができるという仮説を立てました。
オックスフォード大学のチームは、統計ベースのグラフ ネットワークを使用して、タンパク質の機能をその配列からのみ予測することを提案しています。このアプローチは本質的に進化的特徴を特徴づけ、特定の機能を実行する残基の重要性の定量的評価を可能にします。
このメソッドは、進化データから得られた知識を活用して、2 つの積み上げグラフ畳み込みネットワークを駆動します。得られた知識と設計されたネットワーク構造により、タンパク質に機能アノテーションを正確に割り当てることができ、重要なことに、特定の機能に対する各残基の重要性を定量化することができます。
タンパク質の機能注釈用の PhiGnet
PhiGnet メソッドは、統計ベースのグラフ ネットワークを使用して、タンパク質の機能に注釈を付け、配列に基づいて種全体の機能部位を特定します。

進化結合 (EVC) と残基コミュニティ (RC) から知識を吸収するために、研究者はスタックド グラフ畳み込みネットワーク (GCN) を使用したデュアル チャネル アーキテクチャ アプローチを設計しました。このメソッドは、酵素委員会 (EC) 番号や遺伝子オントロジー (GO) 用語 (生物学的プロセス、BP、細胞成分、CC、および分子機能、MF) を含む機能アノテーションをタンパク質に割り当てるように特に設計されています。
タンパク質配列が提供されると、研究では事前トレーニングされた ESM-1b モデルを使用してその埋め込みが導出されます。続いて、埋め込みは、グラフ ノードおよび EVC および RC (グラフ エッジ) としてデュアルスタック GCN の 6 つのグラフ畳み込み層に入力されます。これらの層は、2 つの完全接続 (FC) 層ブロックと連携して両方の GCN からの情報を慎重に処理し、最終的にタンパク質に機能アノテーションを割り当てる実現可能性を評価する確率テンソルを生成します。
さらに、勾配加重クラス活性化マップ (Grad-CAM) メソッドを使用して導出された活性化スコアは、特定の関数の各残基の重要性を評価するために使用されます。このスコアにより、PhiGnet は個々の残基レベルで機能部位を正確に特定できるようになります。
たとえば、セリン-アスパラギン酸リピートを含むプロテイン D (SdrD) の RC を計算すると、機能部位の残基が自然進化を通じて保持されていることが示され、PhiGnet はそのような情報を取得できるため、分析が向上します。構造データがない場合でも、タンパク質の機能を塩基レベルで予測する方法。
タンパク質の機能部位に注釈を付ける
Les prédictions informatiques sont-elles aussi précises que les annotations fonctionnelles déterminées expérimentalement ? Pour répondre à cette question, l’étude a utilisé des scores d’activation pour examiner quantitativement la contribution de chaque acide aminé à la fonction des protéines. Les performances prédictives de PhiGnet ont été évaluées ainsi que l'importance des résidus (leur contribution à la fonction protéique) dans neuf protéines.
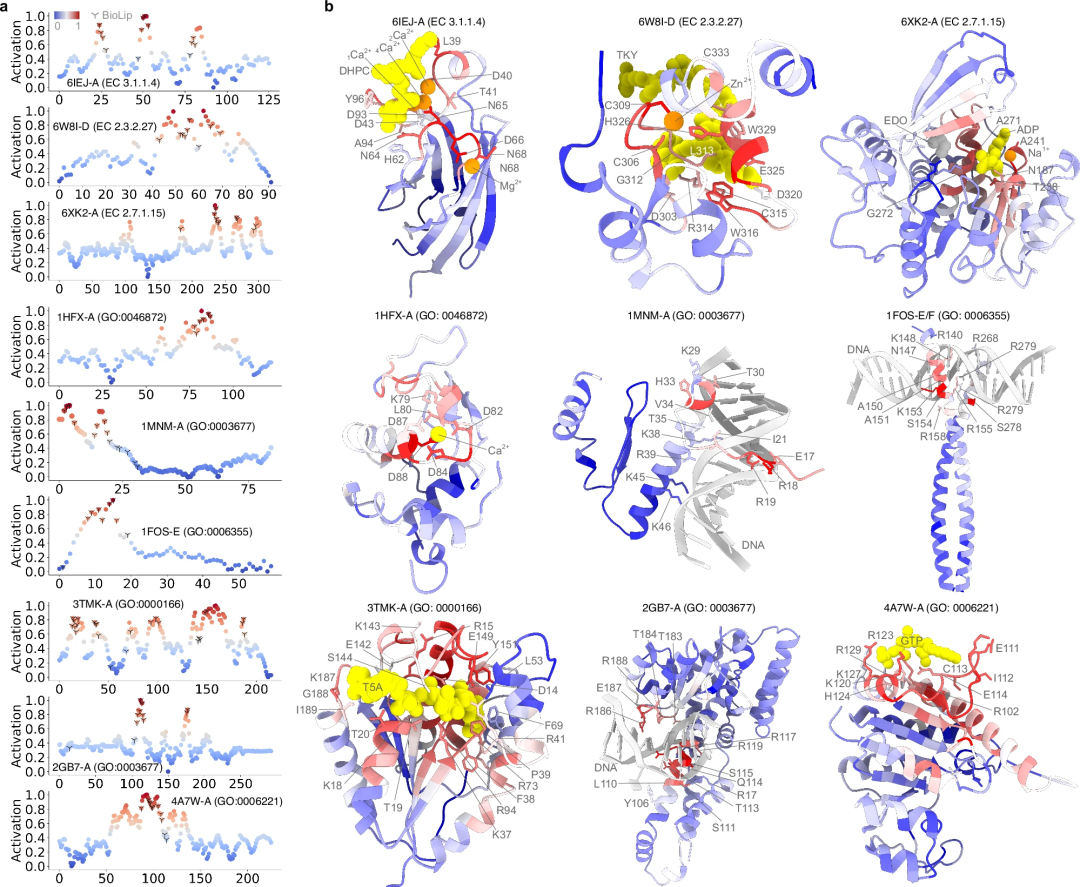
- En calculant le score d'activation pour chaque résidu dans neuf protéines et en les comparant aux résidus déterminés expérimentalement ou par annotation semi-manuelle. PhiGnet a montré une bonne précision (moyenne ⩾ 75 %) dans la prédiction des sites importants au niveau des résidus, en bon accord avec les sites de liaison ligand/ion/ADN réels. PhiGnet identifie avec précision les résidus fonctionnellement importants des protéines avec des scores d'activation élevés.
Surpasse les autres méthodes de pointe
- Pour évaluer les performances de prédiction de PhiGnet, la méthode a été appliquée pour déduire des annotations fonctionnelles (numéros EC et termes GO) de protéines dans deux ensembles de référence. Comparez PhiGnet avec des méthodes de pointe, notamment des méthodes basées sur l'alignement et des méthodes basées sur l'apprentissage en profondeur. Deux mesures de base ont été utilisées à des fins de comparaison, notamment le score Fmax centré sur les protéines et l'aire sous la courbe précision-rappel (AUPR).
Illustration : Comparaison entre différentes méthodes sur les termes GO dans différentes ontologies et numéros EC. (Source : article)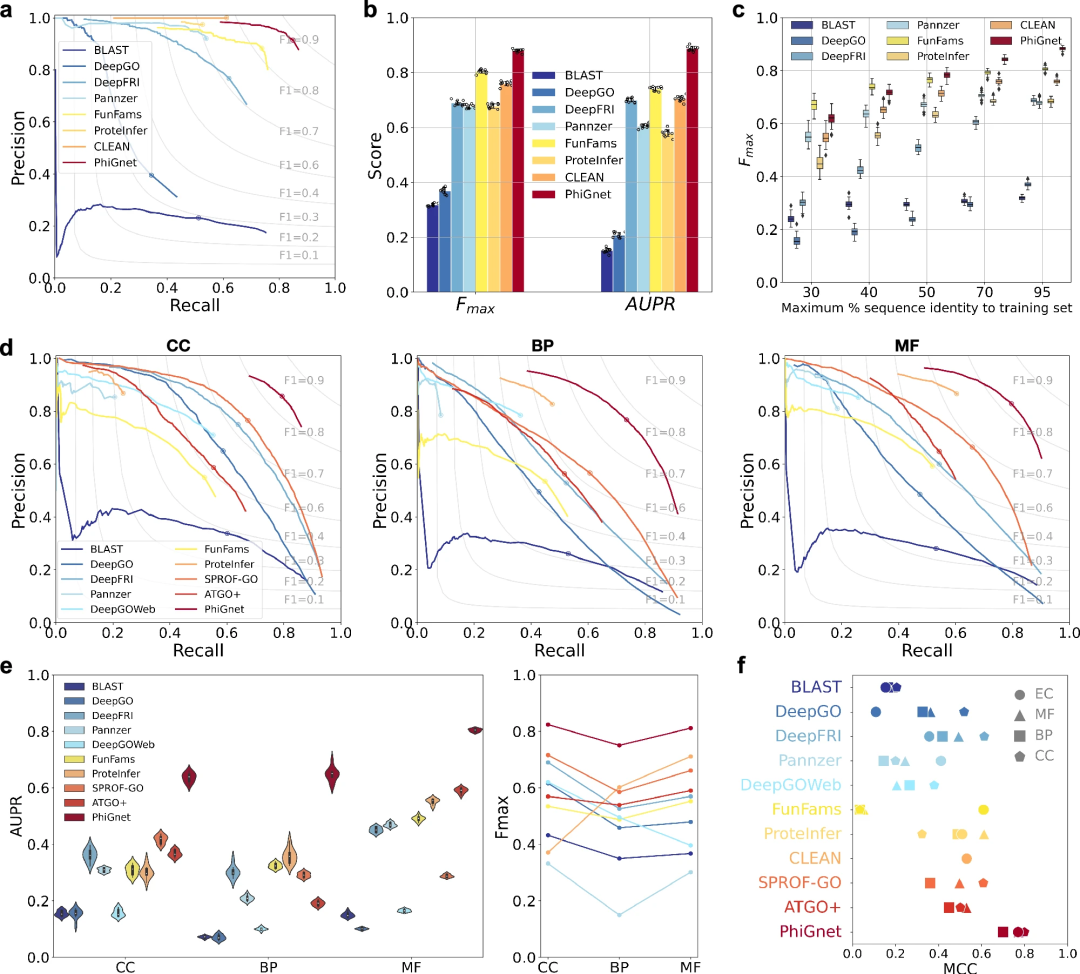
PhiGnet démontre le pouvoir prédictif de l'attribution d'annotations fonctionnelles aux protéines dans deux ensembles de tests. Il atteint des scores AUPR moyens de 0,70 et 0,89 et des scores Fmax de 0,80 et 0,88 pour les termes GO et les numéros EC, respectivement.
Dans l'ensemble, PhiGnet surpasse considérablement toutes les méthodes supervisées et non supervisées sur l'ensemble de données de référence.
De plus, la robustesse de généralisation de PhiGnet a été démontrée pour tester des protéines avec des seuils d'identité de séquence différents de ceux des protéines de l'ensemble d'entraînement. À différents niveaux maximaux d'identité de séquence (30 %, 40 %, 50 %, 70 % et 95 %), PhiGnet a montré de meilleures performances de prédiction à mesure que l'identité de séquence augmentait.
Pilotés par des signatures évolutives
Les données évolutives jouent un rôle important dans PhiGnet et peuvent être utilisées pour prédire les annotations fonctionnelles des protéines et identifier les sites fonctionnels. Tout d’abord, des expériences d’ablation ont été réalisées pour tester la contribution d’EVC/RC à PhiGnet. Les expériences montrent que PhiGnet peut attribuer avec précision des annotations fonctionnelles aux protéines. De plus, PhiGnet utilisant EVC ou RC démontre une forte capacité à apprendre les relations séquence-fonction générales, souvent aussi bien que d'autres méthodes.
Deuxièmement, la capacité de PhiGnet à caractériser des caractéristiques significatives des résidus fonctionnellement pertinents identifiés dans les communautés de résidus a été étudiée plus en détail. Les scores d'activation des résidus ont été calculés pour souligner leur contribution à la fonction des protéines. Notamment, les résidus prédits concordent avec ceux des sites fonctionnels déterminés par des tests expérimentaux et sont mieux identifiés que ceux du RC.
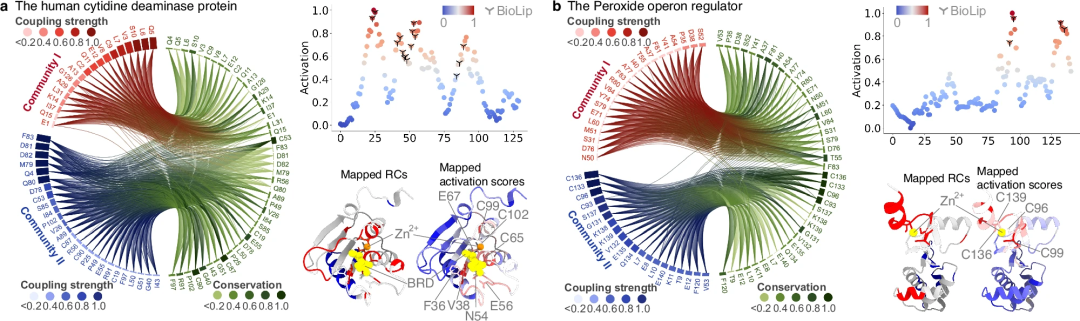
La recherche a montré que les informations évolutives, notamment les informations contenues dans l'homologie à distance, sont suffisantes pour préciser la fonction d'une protéine et caractériser quantitativement les résidus de sites fonctionnels. De plus, l'homologie à distance contient des niveaux d'ordre supérieur de connaissances évolutives par rapport aux niveaux d'informations d'ordre inférieur dans le vecteur évolutif. Dans le même temps, les informations contenues dans Remote Homology jouent un rôle important dans l'amélioration de la capacité de PhiGnet à identifier les sites fonctionnellement pertinents au niveau des résidus.
Succès et limites
En résumé, les meilleures performances de PhiGnet peuvent être attribuées à son utilisation de données évolutives de séquences protéiques et de modèles d'ordre supérieur des données, permettant une compréhension plus profonde et plus précise de la fonction des protéines.
Le principal succès de PhiGnet réside dans l’utilisation de réseaux neuronaux convolutifs de graphiques d’informations statistiques pour faciliter l’apprentissage hiérarchique des données évolutives à partir d’ensembles de données de séquences massifs. Cette approche surpasse considérablement les méthodes supervisées et non supervisées existantes et peut être utilisée pour guider les futures expériences biologiques et cliniques.
Les limites de la méthode PhiGnet incluent le biais/bruit qui se produit dans les familles de protéines avec une faible diversité de séquences. L'incorporation d'informations (co)évolutives dans PhiGnet peut affecter l'identification précise des communautés de résidus, surtout si les informations proviennent de familles de protéines hautement conservées. Bien que l'intégration de connaissances physiquement extraites dans PhiGnet apporte des améliorations significatives par rapport à d'autres approches, des défis importants demeurent dans l'interprétation des mécanismes d'apprentissage dans PhiGnet.
La synergie entre les données évolutives et l'apprentissage automatique ouvrira la voie à la détermination et à l'ingénierie précises des propriétés biophysiques des protéines.
以上がタンパク質機能予測のための新しい SOTA、オックスフォードの上海理工大学などによる統計ベースの AI 手法、Nature サブジャーナルに掲載の詳細内容です。詳細については、PHP 中国語 Web サイトの他の関連記事を参照してください。