
ホームページ >テクノロジー周辺機器 >AI >計算効率は 100 倍以上向上し、非経験的分子動力学計算用の Transformer に基づく大規模なモデルを開発するために Li Jinjin のチームに提出されました。
計算効率は 100 倍以上向上し、非経験的分子動力学計算用の Transformer に基づく大規模なモデルを開発するために Li Jinjin のチームに提出されました。

- WBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBオリジナル
- 2024-06-18 18:02:541173ブラウズ

著者 | Tao Kehao
新世代の高効率材料の開発には、原子や分子の動的挙動の正確なシミュレーションが不可欠です。
しかし、従来の非経験分子動力学 (AIMD) シミュレーションは、当然のことながら高精度の予測機能を提供しますが、高い計算コストと長いシミュレーション時間により、研究の進歩を大きく制限します。
たとえば、100 個の原子を含む材料系の 30 ピコ秒のシミュレーションを構築するには通常数か月かかります。これは、迅速な反復と最適化が必要な新材料の開発にとって大きな課題となります。
この文脈では、このプロセスを大幅に高速化できる人工知能モデルは非常に価値があります。
これらの課題に直面して、上海交通大学人工知能・微細構造研究所 (AIMS-lab) は、T-AIMD と呼ばれる革新的な人工知能モデルを開発しました。
このモデルは、Transformer ベースのネットワーク アーキテクチャを採用しており、計算コストを効果的に削減するだけでなく、あらゆる結晶構造内のあらゆるイオンの挙動を迅速かつ正確に予測します。
このように、T-AIMD モデルは従来の AIMD シミュレーションを 100 倍以上高速化し、材料性能評価プロセスを大幅に高速化します。
さらに、このモデルは混合イオン伝導体の大規模なデータベースの構築に成功し、複数の電池実験でその予測の精度を検証しました。
この方法は、分子動力学モデリング (MD)、生物医薬品分子の結合ターゲット、タンパク質のフォールディング、材料の熱力学プロセス、および機械的特性の計算の分野だけでなく、幅広い応用可能性を持っています。
また、生成人工知能モデルを使用して、より幅広い科学分野の複雑な問題を解決するための新しい方法論も提供します。
T-AIMD の応用の成功は、科学研究と技術革新の促進における人工知能技術の大きな可能性を実証し、将来の新材料研究開発と生物学的設計開発に新たな道を切り開きます。
この研究は「変圧器により固体電解質のイオン輸送挙動の進化と導電性制御が可能」と題され、2024年6月11日に国際的に有名な学術誌「エネルギー貯蔵材料」に掲載されました。
論文の筆頭著者は上海交通大学人工知能・微細構造研究室の博士課程学生Tao Kehao氏、責任著者は研究室所長のLi Jinjin教授です。

記事リンク: https://www.sciencedirect.com/science/article/pii/S2405829724003829
人工知能の分野では、Transformer モデルは複雑なシーケンス データを処理するための推奨フレームワークとなっています。 。
このモデルは、大規模なデータから深いパターンや関連性を学習することに特に優れているため、言語処理、画像認識、さまざまな予測タスクで広く使用されています。
それにもかかわらず、Transformer の可能性は、材料科学、特に非経験的分子動力学 (AIMD) シミュレーションのアプリケーションではまだ十分に活用されていません。
従来の AIMD シミュレーションは、原子や分子の動的挙動を正確にシミュレートできるため、材料科学において非常に重要です。ただし、そのようなシミュレーションは繰り返しの計算と高価な実験に依存することが多く、時間がかかるだけでなくコストもかかります。
このような課題に直面すると、大量の配列データを迅速に抽出して処理できるインテリジェントなモデルが特に重要になります。
この需要に応えて、上海交通大学の AIMS 研究室チームによって開発された T-AIMD モデルは、Transformer ネットワーク アーキテクチャを使用して、AIMD シミュレーションの速度と精度を大幅に向上させています。
この新しいモデルは、計算コストを大幅に削減しながら、さまざまな条件下での原子や分子の挙動を迅速かつ正確に分析および予測できます。
従来の AIMD 手法と比較して、T-AIMD はシミュレーション速度を 100 倍以上向上させることができ、同時に高い予測精度を維持し、材料開発サイクルを大幅に短縮します。
これは、材料科学分野の研究に新しいツールを提供するだけでなく、高性能コンピューティングタスクにおける AI の応用可能性を実証し、将来の科学探査に新たな可能性を開きます。
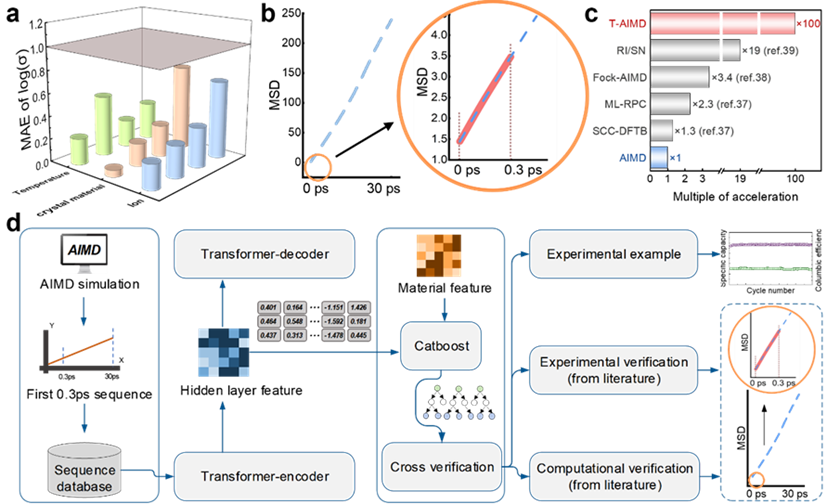
イラスト: T-AIMD の予測結果とワークフロー図。 (出典: 論文)
固体電解質におけるイオン輸送挙動を予測する問題を解く例を考えてみましょう。モデルは電解質内のイオンの拡散シーケンスを学習することで、将来の状態での挙動を予測することができ、材料特性の評価プロセスを大幅に加速します。
さらに、T-AIMD モデルにはマルチソース材料記述子も組み込まれており、複雑な材料システムの処理におけるアプリケーション機能が強化され、単一イオン種の挙動を予測するだけでなく、マルチイオンシステムにおける相互作用も処理できるようになります。アクションと複雑なダイナミクスの問題。
この新しいトランスベースの方法は、固体電解質の開発に新しい視点とツールを提供し、材料科学の分野で新しい研究と応用の可能性を開くことが期待されています。
T-AIMDの仕組みについて
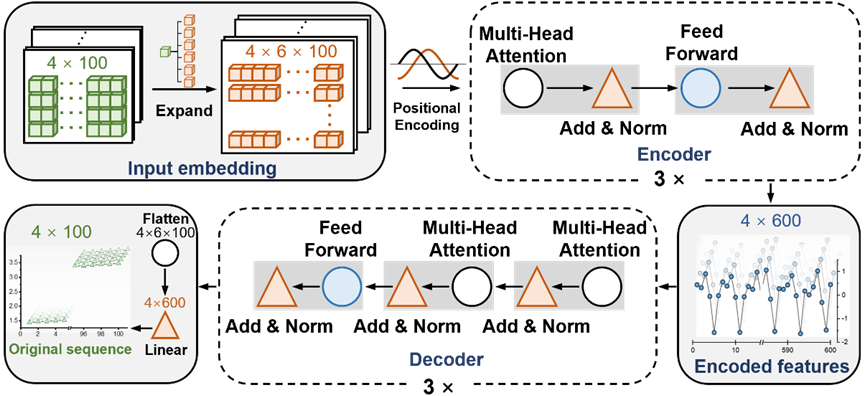
イラスト: T-AIMDのネットワークアーキテクチャ図。 (出典: 論文)
T-AIMD (Transformer-based Ab Initio Molecular Dynamics) は、非経験分子動力学 (AIMD) シミュレーションと Transformer ディープラーニング アーキテクチャを組み合わせたモデルで、固体電解質材料におけるイオン輸送速度の向上を目的としています。動作特性の予測の精度。このモデルの動作原理は、次の主要なステップに分けることができます:
1. データの準備と前処理
T-AIMD はまず、従来の AIMD シミュレーションから得られる材料のイオン拡散データを収集します。これらのシミュレーションによって生成されたデータには、電解質を通るイオンの動きを記録する時系列データが含まれます。これらのシーケンス データは前処理され、機械学習モデルへの入力として適切な形式に変換されます。
2. 特徴抽出
T-AIMD は、Transformer モデルのエンコーダー部分を使用して、シーケンス データから主要な特徴を抽出できます。このプロセスでは、モデルは自己注意メカニズムを通じてシーケンス内の長距離依存関係を捕捉します。これは複雑なイオン ダイナミクスを理解するために重要です。
3. シーケンスの学習と予測
特徴抽出後、Transformer モデルのデコーダー部分を使用して、エンコードされた特徴に基づいてシーケンス予測を実行します。このステップでは、モデルはイオンの将来の挙動を予測できるだけでなく、さまざまな温度や圧力などのさまざまな条件下でのイオンの潜在的な挙動を分析することもできます。さらに、モデルはこれらの学習された特徴を通じて、材料のイオン伝導率などの重要な性能指標を予測できます。
4. マルチソース材料記述子の統合
T-AIMD は、結晶構造、イオン種、電子特性などのさまざまなソースからの材料記述子を組み合わせて、モデルがより包括的に材料を理解して予測するのに役立ちます。プロパティ。この統合されたアプローチにより、モデルの多用途性とさまざまな材料システムへの適応性が向上します。
5. モデルの検証と応用
開発したモデルは、実験データや他の計算手法と比較して、予測精度を検証する必要があります。検証が成功した後は、T-AIMD を使用して新しいターゲット材料を迅速にスクリーニングおよび最適化することができるため、開発サイクルが大幅に短縮され、コストが削減されます。
T-AIMD の堅牢なパフォーマンスについて
T-AIMD モデルの堅牢なパフォーマンスは主に次の側面に反映されます:
1. 精度
T-AIMD モデルは、Transformer アーキテクチャを統合しています。複雑な動的動作を学習および予測する能力が大幅に強化されています。 AIMD シミュレーションの高速化に関しては、T-AIMD は従来の方法よりも高い精度を示します。これは、モデルがより短い時間でより長い時間スケールでイオンの挙動を正確に予測できる深層学習技術の適用によるものです。
2. 計算効率
計算効率の点では、T-AIMD は従来の AIMD 手法よりも大幅に優れています。従来の AIMD シミュレーションはイオン拡散のシミュレーションに多くの時間を要しますが、T-AIMD は計算プロセスを最適化することで高性能コンピューティング リソースへの依存を大幅に軽減し、シミュレーション時間を数か月から数日または数時間に短縮します。
3. 汎用性と柔軟性
T-AIMD は、従来の機械学習モデル (サポート ベクター マシンやデシジョン ツリーなど) よりも複雑なデータ構造と大規模なデータ セットを処理できます。このモデルは、さまざまな種類の材料に適応し、温度や圧力の変化など、さまざまな環境条件下での挙動を効果的に予測できます。
4. モデルの堅牢性
T-AIMD は、ノイズや不確実性のあるデータを扱う場合に高い堅牢性を示します。比較実験では、T-AIMD はデータにわずかな偏りがある場合でも高い予測精度を維持できますが、これは他の単純な機械学習モデルでは達成することが困難です。
5. スケーラビリティと適応性
T-AIMD モデルのアーキテクチャにより、変化する研究ニーズや新しい科学的発見に適応するための柔軟な調整と最適化が可能になります。この拡張性により、T-AIMD は今後の研究でも重要な役割を果たし続けることが可能となり、その用途は固体電解質を超えて他のエネルギー材料や複雑な化学系の研究にまで広がります。
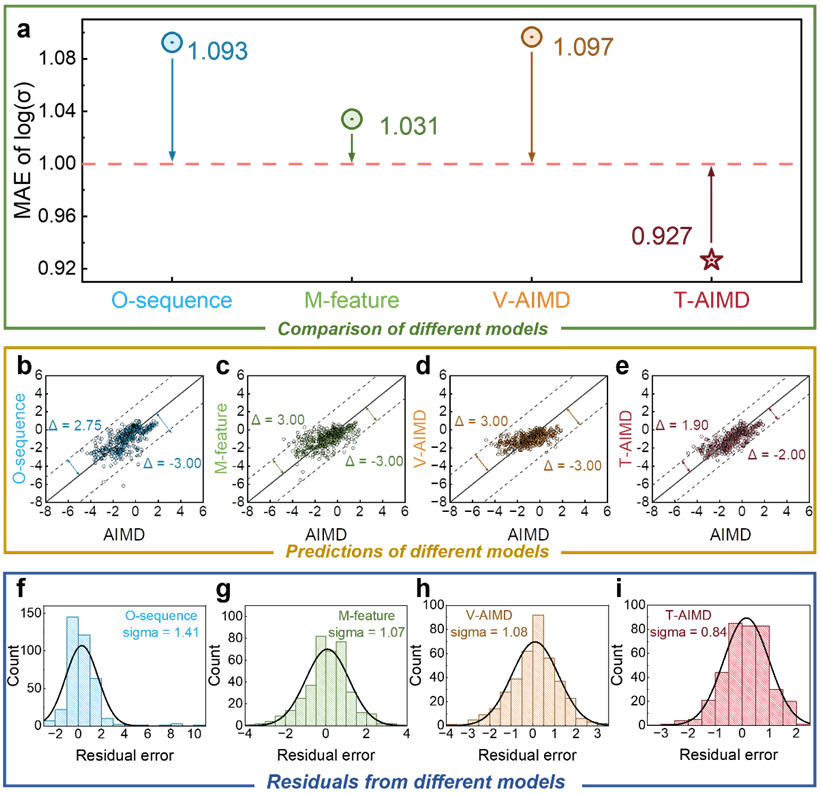
要約すると、T-AIMD フレームワークに基づいて、分子動力学のシミュレーション効率を大幅に加速することができ、効率が 1000 倍、10000 倍、あるいはそれ以上向上し、シミュレーションにかかる時間を大幅に節約できます。材料の製造と生物学的設計には時間コストがかかります。
T-AIMD モデルは、いくつかの重要な側面において従来の AIMD シミュレーションやその他の機械学習手法を上回っており、本文中に挙げられた例は、固体電解質の研究開発におけるその強力な可能性と応用の見通しを示しています。
T-AIMD の実用性はそれをはるかに超えています。このモデルの能力と柔軟性により、材料科学の多くの分野に広く適用できます。
将来的には、半導体、金属、高分子材料など、他の種類の材料におけるイオンや分子の挙動を予測するために使用されることが期待されています。
さらに、T-AIMD モデルの機能は単一イオン種の挙動の予測に限定されず、マルチイオン系における複雑な相互作用やダイナミクス問題も処理できるため、新しい設計に役立ちます。既存の材料の性能を向上させ、非常に高い実用価値を持っています。
以上が計算効率は 100 倍以上向上し、非経験的分子動力学計算用の Transformer に基づく大規模なモデルを開発するために Li Jinjin のチームに提出されました。の詳細内容です。詳細については、PHP 中国語 Web サイトの他の関連記事を参照してください。