
Maison >Périphériques technologiques >IA >Précision > 98 %, GPT basé sur la densité électronique est utilisé dans la recherche chimique, publié dans la sous-journal Nature
Précision > 98 %, GPT basé sur la densité électronique est utilisé dans la recherche chimique, publié dans la sous-journal Nature

- PHPzavant
- 2024-03-27 14:16:121271parcourir
Editeur | Violet
L'espace chimique des molécules synthétisées est très large. Une exploration efficace de ce domaine nécessite la confiance sur le criblage informatique Des technologies, telles que l'apprentissage profond, pour découvrir rapidement une variété de composés intéressants
La conversion des structures moléculaires en représentations numériques et le développement d'algorithmes correspondants pour générer de nouvelles structures moléculaires sont la clé de la découverte chimique
Récemment, l'Université de Glasgow, Royaume-Uni L'équipe de recherche a proposé un modèle d'apprentissage automatique basé sur la formation à la densité électronique pour générer des classeurs hôte-invité. Ce modèle est capable de lire des données au format SMILES (Simplified Molecular Linear Input Specification) avec une précision allant jusqu'à 98 %. une précision de 98 % Description complète des molécules dans un espace bidimensionnel
Générez une représentation tridimensionnelle de la densité électronique et du potentiel électrostatique du système hôte-invité via un auto-encodeur variationnel, puis optimisez la génération de l'invité. par descente de gradient. Enfin, utilisez le transformateur pour convertir l'invité en SMILES. Une représentation et une transformation efficaces des structures invitées ont été appliquées avec succès aux systèmes hôtes moléculaires établis, au cucurbituril et aux cages métallo-organiques, ce qui a permis la découverte de 9. invités CB[6] précédemment validés et 7 objets non signalés, et découvert 4 GPT à 98 % non signalés basés sur la densité électronique pour la recherche chimique, publiés dans la sous-journal Nature " />Objets.
La recherche s'intitulait « GPT basé sur la densité électronique pour l'optimisation et la suggestion de classeurs hôte-invité » et a été publiée dans « Nature Computational Science » le 8 mars 2024. 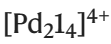
Lien papier : https://www.nature.com/articles/s43588-024-00602-x 
Un modèle d'apprentissage automatique formé sur la densité électronique
Ici, il est démontré que la représentation d'une molécule hôte sous la forme d'un volume 3D (c'est-à-dire une densité électronique modifiée avec un potentiel électrostatique) permet une découverte assistée par ordinateur des invités de l'hôte. pas besoin de comprendre le système hôte-invité au-delà de la structure chimique du sujet.
Au cours du processus, les chercheurs ont construit un modèle Transformer qui peut être entraîné pour convertir efficacement des descripteurs moléculaires volumétriques 3D en représentations SMILES, générant ainsi des structures moléculaires utilisables par les chimistes professionnels. L'étude a également révélé que les molécules peuvent être représentées efficacement sous forme de volumes 3D en modifiant leur densité électronique avec des données de potentiel électrostatique, et que ces deux caractéristiques sont suffisantes pour optimiser la forme et la charge du volume entre les descripteurs 3D en utilisant un schéma d'échantillonnage autorégressif. Molécules invitées interagissez pour découvrir l’hôte.98 %, le GPT basé sur la densité électronique est utilisé dans la recherche chimique, publié dans le sous-journal Nature" />
![准确率 ></section>Le modèle Transformer prédit parfaitement sa représentation SMILES avec une précision de 98,125 %. La précision de prédiction pour un seul jeton est de 99,114 %. modèle génératif <section></section> <p>Aperçu du workflow</p><p>La découverte assistée par ordinateur du cucurbituril CB[6] et la validation expérimentale des cages métallo-organiques <p> nécessitent un flux de travail à deux niveaux. Tout d’abord, un flux de travail in silico a été conçu pour générer des bibliothèques virtuelles de molécules invitées potentielles pour les deux hôtes. Un flux de travail in vitro a ensuite été établi, comprenant la sélection des candidats invités les plus prometteurs de ces bibliothèques virtuelles par des chimistes experts pour des tests expérimentaux. <img src=](https://img.php.cn/upload/article/000/000/164/171152017578536.png)
98 %, le GPT basé sur la densité électronique est utilisé dans la recherche chimique, publié dans le sous-journal Nature" />![准确率 ></p>Illustration : Découverte de nouvelles molécules invitées grâce à la représentation volumique de la densité électronique. (Source : article) <section></section>CB[ 6] et <p> La génération informatique de molécules invitées est réalisée grâce au flux de travail illustré dans la figure ci-dessus, <img src=](https://img.php.cn/upload/article/000/000/164/171152017597952.png) Le flux de travail comprend les étapes suivantes :
Le flux de travail comprend les étapes suivantes :
grâce à des simulations informatiques, un flux de travail in vitro a été établi pour tester expérimentalement les candidats les plus prometteurs. 
générés grâce à son flux de travail informatique ont été classés par des experts en chimie pour des tests expérimentaux. Les invités prometteurs à tester sont sélectionnés en fonction de leur similarité structurelle avec les invités connus de CB[6] ou , de l'intuition des chimistes professionnels et de leur disponibilité commerciale.
(2) Utilisation directe du  Méthode de titrage pour déterminer l'affinité du CB[6] ou . Il est à noter que l'invité généré dans l'ordinateur Mélanges contenant des molécules connues auparavant pour se lier à (ou étroitement liées) à des hôtes et des molécules qui défient l'intuition des experts
Méthode de titrage pour déterminer l'affinité du CB[6] ou . Il est à noter que l'invité généré dans l'ordinateur Mélanges contenant des molécules connues auparavant pour se lier à (ou étroitement liées) à des hôtes et des molécules qui défient l'intuition des experts
Validation expérimentale de deux systèmes hôte-invité communs
Les chercheurs ont validé expérimentalement leur flux de travail pour deux hôtes communs. -systèmes invités : le cucurbituril (CB[n]) et les cages métallo-organiques sont devenus des objets vérifiés dans la littérature et non signalés. L'algorithme a généré 9 objets précédemment connus pour CB[6]. Sept nouveaux invités potentiels de CB[6] ont également été identifiés. et l'affinité de CB[6] pour ces nouveaux invités a été évaluée par
titrage direct en HCO2H/H2O 1:1v/vDans les 7 cas, un ensemble de signaux est observé pour le système hôte-invité, indiquant que le système hôte-invité. Le système subit un échange rapide sur l'échelle de temps RMN. Après la complexation, la résonance de la chaîne aliphatique des molécules invitées se déplace vers le haut, indiquant qu'elles sont encapsulées dans la cavité CB[6] trouvée  98 %, GPT basé sur la densité électronique. Chemical Research, publié dans Nature Sub-Journal" /> La constante d'association avec CB[6] suit la tendance précédemment établie, allant de 13,5 M^− 1 à 5 470 M^−1.
98 %, GPT basé sur la densité électronique. Chemical Research, publié dans Nature Sub-Journal" /> La constante d'association avec CB[6] suit la tendance précédemment établie, allant de 13,5 M^− 1 à 5 470 M^−1.

Illustration : optimisation de CB[6] et objets précédemment connus et objets optimisés de 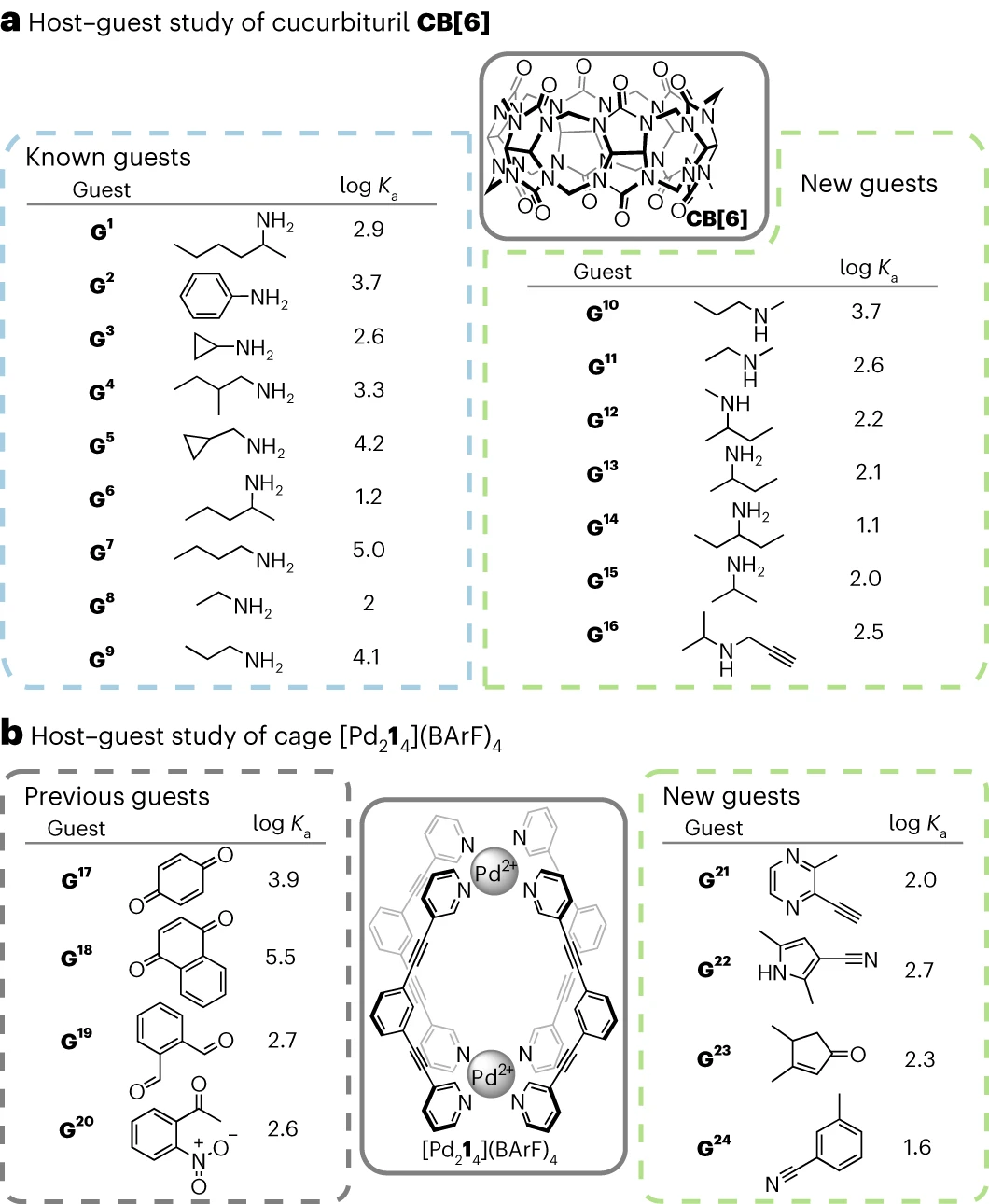 , l'algorithme d'optimisation a généré uniquement des molécules invitées inconnues, et la force de liaison entre quatre invités potentiels non signalés et [Pd214](BArF)4 a été testée par titrage direct
, l'algorithme d'optimisation a généré uniquement des molécules invitées inconnues, et la force de liaison entre quatre invités potentiels non signalés et [Pd214](BArF)4 a été testée par titrage direct
Alors que la recherche s'est concentrée sur l'utilisation de la notation SMILES pour représenter des molécules, d'autres formats similaires tels que les chaînes intégrées auto-référentielles (SELFIES) ont également été testés.
Bien que l'ensemble de données QM9 contienne des molécules parfaitement dimensionnées pour devenir des invités pour des hôtes tels que CB[6], une limitation rencontrée par cette étude est que les cages métallo-organiques ont des cavités plus grandes et nécessitent des molécules invitées plus grosses. Dans les études futures, des ensembles de données contenant des molécules plus grosses, tels que l'ensemble de données GDB-17, seront utilisés.
Après cela, « Notre objectif est d'intégrer la sélection de nouveaux ligands dans le processus de génération, de synthétiser de manière autonome des molécules sur des plateformes de synthèse automatisées (telles que les robots Chemputer), de boucler la boucle entre optimisation et tests et de créer un environnement cyber-physique fermé. Système de boucle."
Ce qui précède est le contenu détaillé de. pour plus d'informations, suivez d'autres articles connexes sur le site Web de PHP en chinois!