
Maison >Périphériques technologiques >IA >L'IA ne s'apprend pas ! Une nouvelle recherche révèle des moyens de déchiffrer la boîte noire de l'intelligence artificielle
L'IA ne s'apprend pas ! Une nouvelle recherche révèle des moyens de déchiffrer la boîte noire de l'intelligence artificielle

- WBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBavant
- 2024-01-16 21:30:31986parcourir
L'intelligence artificielle (IA) se développe rapidement, mais pour les humains, les modèles puissants sont une « boîte noire ».
Nous ne comprenons pas le fonctionnement interne du modèle et le processus par lequel il parvient à ses conclusions.
Cependant, récemment, le professeur Jürgen Bajorath, expert en cheminformatique à l'Université de Bonn, et son équipe ont réalisé une avancée majeure.
Ils ont conçu une technique qui révèle le fonctionnement de certains systèmes d'intelligence artificielle utilisés dans la recherche sur les médicaments.
La recherche montre que les modèles d'intelligence artificielle prédisent l'efficacité des médicaments principalement en rappelant des données existantes, plutôt qu'en apprenant des interactions chimiques spécifiques.
——En d'autres termes, les prédictions de l'IA reposent uniquement sur la reconstitution de souvenirs, et l'apprentissage automatique n'apprend pas réellement !
Les résultats de leurs recherches ont été récemment publiés dans la revue Nature Machine Intelligence.

Adresse papier : https://www.nature.com/articles/s42256-023-00756-9
Dans le domaine de la médecine, les chercheurs recherchent fébrilement des substances actives efficaces pour lutter contre les maladies. —Quelles molécules médicamenteuses sont les plus efficaces ?
Habituellement, ces molécules (composés) efficaces sont liées à des protéines, qui agissent comme des enzymes ou des récepteurs qui déclenchent des chaînes d'action physiologiques spécifiques.
Dans des cas particuliers, certaines molécules sont également chargées de bloquer les réactions indésirables dans l'organisme, comme les réponses inflammatoires excessives.
Le nombre de composés possibles est énorme et trouver celui qui fonctionne, c'est comme chercher une aiguille dans une botte de foin.
Les chercheurs ont donc d'abord utilisé des modèles d'IA pour prédire quelles molécules s'ancreraient le mieux et se lieraient fortement à leurs protéines cibles respectives. Ces candidats médicaments sont ensuite examinés plus en détail dans le cadre d’études expérimentales.
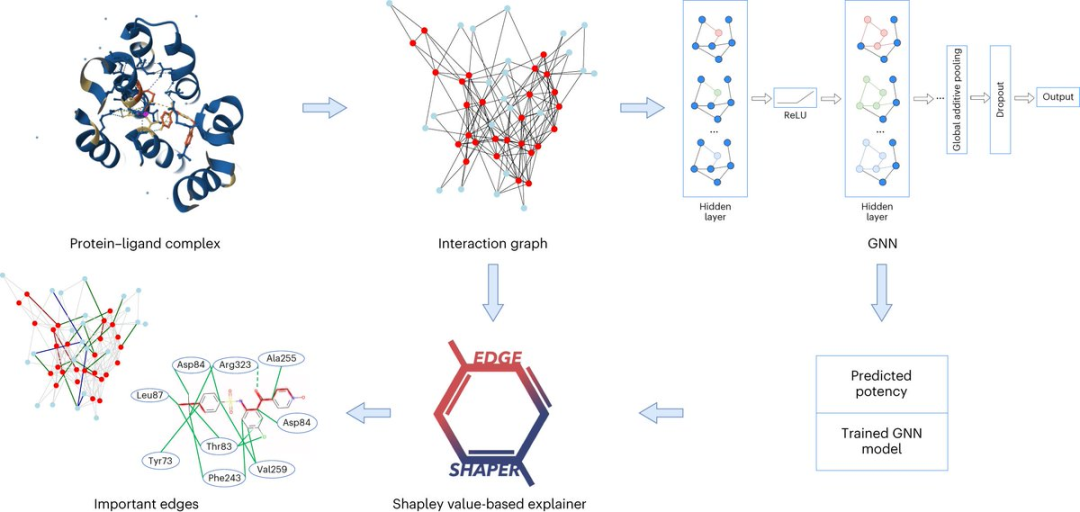
Depuis le développement de l'intelligence artificielle, la recherche sur la découverte de médicaments a de plus en plus adopté les technologies liées à l'IA.
Par exemple, le réseau neuronal graphique (GNN) convient pour prédire la force de liaison d'une certaine molécule à une protéine cible.
Un graphe se compose de nœuds représentant des objets et d'arêtes représentant les relations entre les nœuds. Dans la représentation graphique d'un complexe protéine-ligand, les bords du graphique relient des nœuds protéine ou ligand, représentant la structure d'une substance ou l'interaction entre une protéine et un ligand.
Le modèle GNN utilise des cartes d'interaction protéine-ligand extraites de structures radiographiques pour prédire les affinités des ligands.
Le professeur Jürgen Bajorath a déclaré que le modèle GNN est pour nous comme une boîte noire et que nous n'avons aucun moyen de savoir comment il dérive ses prédictions.

Le professeur Jürgen Bajorath travaille à l'Institut LIMES de l'Université de Bonn, au Centre international de technologie de l'information de Bonn-Aix-la-Chapelle (Bonn-Aachen International Center for Information Technology) et à l'Institut Lamarr pour l'apprentissage automatique et l'intelligence artificielle (Institut Lamarr pour l'apprentissage automatique et l'intelligence artificielle).
Comment fonctionne l'intelligence artificielle ?
Des chercheurs du département d'informatique chimique de l'université de Bonn, en collaboration avec des collègues de l'université Sapienza de Rome, ont analysé en détail si le réseau neuronal graphique avait réellement appris l'interaction entre la protéine et le ligand.
Les chercheurs ont analysé un total de six architectures GNN différentes à l'aide de leur méthode "EdgeSHAPer" spécialement développée.
Le programme EdgeSHAPer peut déterminer si le GNN a appris les interactions les plus importantes entre les composés et les protéines, ou fait des prédictions par d'autres méthodes.
Les scientifiques ont formé six GNN à l'aide de graphiques extraits des structures de complexes protéine-ligand - où le mode d'action du composé et la force de sa liaison à la protéine cible étaient connus.
Ensuite, testez le GNN formé sur d'autres composés et utilisez EdgeSHAPer pour analyser comment le GNN produit des prédictions.
« Si les GNN se comportent comme prévu, ils doivent apprendre les interactions entre les composés et les protéines cibles et faire des prédictions en priorisant les interactions spécifiques. »
Cependant, selon l’analyse de l’équipe de recherche, six GNN n’ont pratiquement pas réussi à y parvenir. La plupart des GNN n’apprennent que certaines interactions protéine-médicament, en se concentrant principalement sur les ligands.
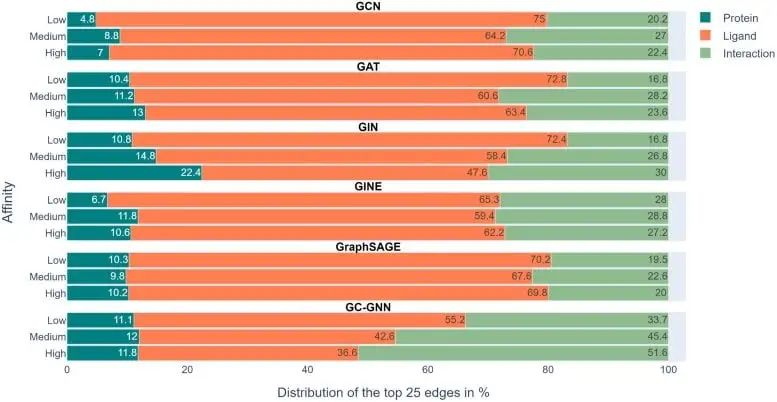
La figure ci-dessus montre les résultats expérimentaux dans 6 GNN. La barre d'échelle de couleurs représente la proportion moyenne de protéines, de ligands et d'interactions dans les 25 bords supérieurs de chaque prédiction déterminée par EdgeSHAPer.
Nous pouvons voir que l'interaction représentée par le vert est ce que le modèle doit apprendre, mais sa proportion dans l'ensemble de l'expérience n'est pas élevée et la barre orange représentant le ligand représente la plus grande proportion.
Pour prédire la force de liaison d'une molécule à une protéine cible, les modèles « se souviennent » principalement des molécules chimiquement similaires et de leurs données de liaison qu'ils ont rencontrées au cours de l'entraînement, quelle que soit la protéine cible. Ces similitudes chimiques mémorisées déterminent essentiellement la prédiction.
Cela n'est pas sans rappeler "l'effet Hans intelligent" - tout comme le cheval qui semble savoir compter, mais qui compte en réalité en fonction des nuances des expressions faciales et des gestes de son compagnon, pour en déduire les résultats attendus.
Cela peut signifier que la soi-disant « capacité d'apprentissage » du GNN peut être intenable et que les prédictions du modèle sont largement surestimées car les connaissances chimiques et des méthodes plus simples peuvent être utilisées pour effectuer des prédictions de même qualité.
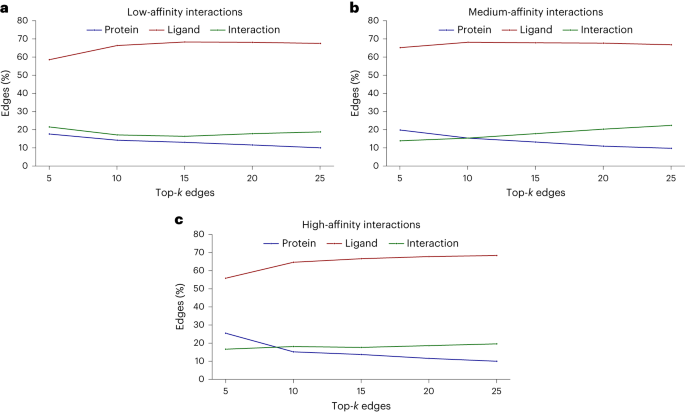
Cependant, un autre phénomène a également été découvert dans l'étude : lorsque la puissance du composé testé augmente, le modèle a tendance à apprendre davantage d'interactions.
Peut-être qu'en modifiant les techniques de représentation et de formation, ces GNN pourront être encore améliorés dans la direction souhaitée. Cependant, l’hypothèse selon laquelle les grandeurs physiques peuvent être apprises à partir de graphiques moléculaires doit généralement être traitée avec prudence.
"L'intelligence artificielle n'est pas de la magie noire."
Ce qui précède est le contenu détaillé de. pour plus d'informations, suivez d'autres articles connexes sur le site Web de PHP en chinois!