
Maison >Périphériques technologiques >IA >Annonce du prix Gordon Bell 2023 : la simulation de matériaux « Quantum Level Accuracy » de Frontier Supercomputer remporte le prix
Annonce du prix Gordon Bell 2023 : la simulation de matériaux « Quantum Level Accuracy » de Frontier Supercomputer remporte le prix

- WBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBavant
- 2023-11-18 12:25:55801parcourir

Éditeurs | Zenan, Du Wei
Le prix ACM Gordon Bell a été créé en 1987 et décerné par l'American Computer Society. Il est connu sous le nom de « prix Nobel » dans le domaine du calcul intensif. Ce prix est décerné chaque année pour reconnaître l'excellence en calcul haute performance. Le prix est de 10 000 $, offert par Gordon Bell, un pionnier du calcul parallèle et haute performance.
Lors de la récente conférence mondiale sur le calcul intensif SC23, une équipe internationale de 8 membres de chercheurs américains et indiens a remporté le prix ACM Gordon Bell 2023 pour leur succès. dans la réalisation de simulations de précision quantique à grande échelle des matériaux. Le nom de ce projet est « Modélisation quantique de matériaux à grande échelle : simulations ab initio de quasicristaux et défauts étendus par interaction dans les alliages métalliques »
Les membres de l'équipe viennent de l'Université du Michigan, du Laboratoire national d'Oak Ridge et de l'Indian Institute. des sciences (Bangalore).

Membre de l'équipe primé.
Auparavant, le prix Gordon Bell 2021 avait été décerné à une équipe chinoise d'application de calcul intensif composée de 14 membres, comprenant des membres du laboratoire Zhijiang et du Centre national de calcul intensif Wuxi, de l'Université Tsinghua et du Centre de recherche en sciences quantiques de Shanghai, en reconnaissance de la nouvelle génération de l'équipe. de recherche basée sur l'application du supercalculateur Sunway de notre pays "Simulation en temps réel de circuits aléatoires quantiques à très grande échelle". À l'avenir, l'équipe chinoise chargée des applications du supercalcul a également remporté le prix Gordon Bell pendant deux années consécutives, en 2016 et 2017.
Aperçu de la recherche
Contenu réécrit : Nous avons appris que la dynamique moléculaire est une méthode d’utilisation de simulations informatiques pour mieux comprendre les processus par lesquels les atomes et les molécules se déplacent au sein d’un système. Une branche de la dynamique moléculaire est "Ab initio", une technique qui s'est avérée très efficace pour résoudre des problèmes importants en physique et en chimie, tels qu'une meilleure compréhension des mécanismes microscopiques, l'obtention de nouvelles connaissances en science des matériaux, la vérification des données expérimentales et plus encore.

Adresse papier :https://dl.acm.org/doi/pdf/10.1145/3581784.3627037
Ce projet est dirigé par Vikram Gavini, professeur de génie mécanique, de science et d'ingénierie des matériaux à l'Université du Michigan La recherche a utilisé le Frontier (supercalculateur HPE Cray EX 1,14 exaflop) du Laboratoire national d'Oak Ridge du Département américain de l'énergie pour effectuer des simulations en utilisant une approche des principes premiers via l'équation de Schrödinger, qui décrit les systèmes microscopiques, y compris leurs propriétés probabilistes. Selon les rapports, les résultats peuvent être utilisés pour aider à concevoir des matériaux candidats pour de nouveaux alliages et promouvoir d'autres efforts de conception informatique tels que la découverte de médicaments.
L'équipe de Gavini a utilisé un cadre informatique intégré sur les supercalculateurs Frontier et Summit pour simuler des dislocations, ou défauts, dans un système de magnésium composé de près de 75 000 atomes. Les alliages de magnésium sont des candidats prometteurs pour les alliages légers, mais des lacunes mal alignées dans la structure atomique du magnésium peuvent entraîner une fragilité et des fissures. Comprendre les dislocations dans les alliages de magnésium pourrait fournir à l'industrie des alliages plus légers et plus flexibles
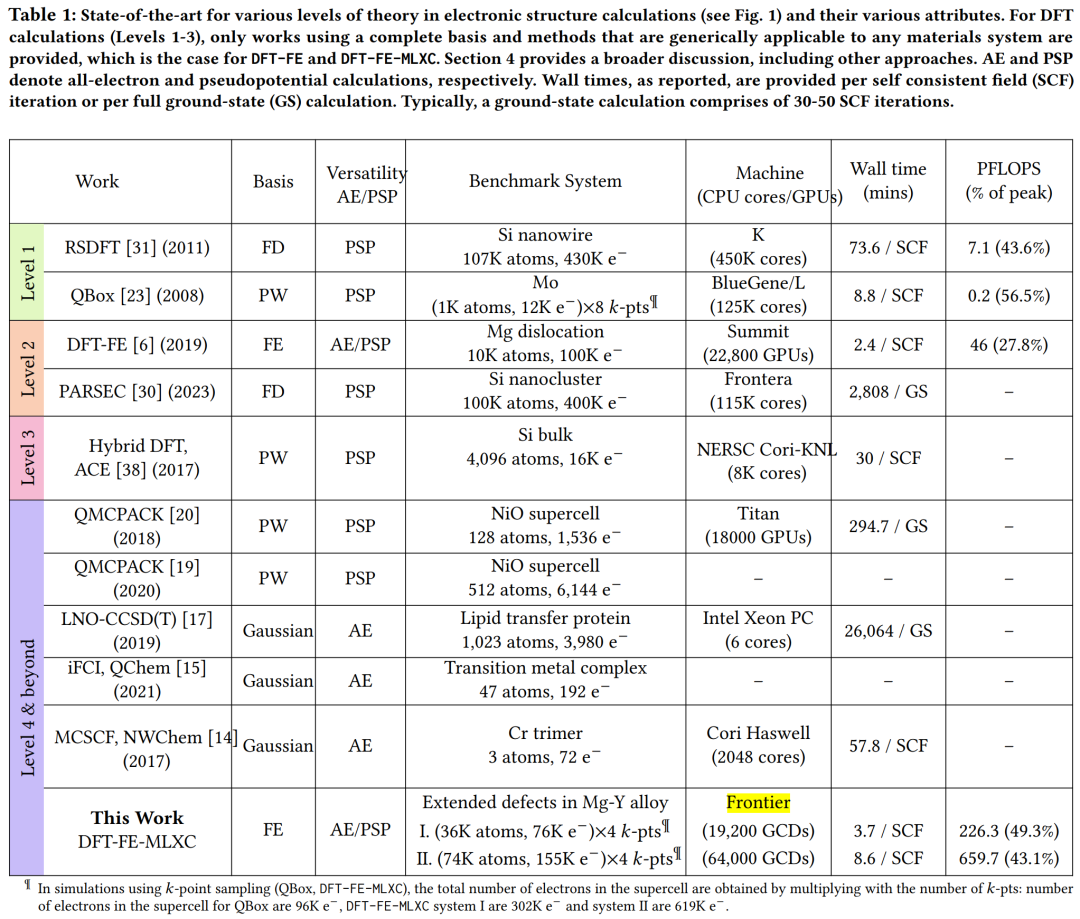
Cet article est comparé à des travaux antérieurs
L'équipe utilise également le supercalculateur Perlmutter du National Energy Research Center for Scientific Computing pour étudier la stabilité des quasi-cristaux ( une structure ordonnée mais non périodique) dans les alliages ytterbium-cadmium.
Ces calculs s'appuient sur la théorie fonctionnelle de la densité, une méthode mécanique quantique de calcul de la structure atomique et électronique des matériaux, et utilisent l'apprentissage automatique pour approcher le haut niveau de précision des calculs quantiques à N corps. Ils ont utilisé les 8 000 nœuds de Frontier, avec une puissance de calcul maximale de 659,7 pétaflops. « Alors que nous nous efforçons d'atteindre une plus grande précision, le nombre de systèmes informatiques disponibles a considérablement diminué », a déclaré Gavini. "Nous utilisons les résultats de calculs quantiques à N corps sur des systèmes plus petits et utilisons l'apprentissage automatique pour déduire des relations constitutives universelles pour les électrons, qui peuvent être utilisées dans des calculs de théorie fonctionnelle à plus grande densité. En combinant ces méthodes, nous sommes en mesure d'utiliser des outils comme Frontier The avantage d'une si grande machine, tout en se rapprochant de la précision quantique. Cet article vise à décrire une méthode pour y parvenir grâce à des simulations de matériaux à grande échelle avec une précision quantique. Les derniers résultats de recherche de l'équipe Frontier sont les premiers depuis dix ans. dans nos efforts. Avant cela, une étude réalisée en 2019 avait utilisé Summit pour simuler plus de 10 000 atomes de magnésium et avait été nominée pour le Gordon Bell Award
Le processus de production des alliages implique la fusion et le mélange des métaux. Des défauts peuvent apparaître lors du processus de solidification et avoir un impact positif ou négatif sur les propriétés du matériau. La structure atomique du matériau joue un rôle crucial dans le comportement de ces défauts linéaires, souvent appelés dislocations.
Les métaux malléables comme l'aluminium bénéficient d'une structure atomique, permettant au métal de s'adapter aux dislocations et à leur mouvement. La structure atomique du magnésium ne peut pas facilement s'adapter aux dislocations, ce qui la rend plus fragile par nature.
Dans de bonnes circonstances, ces défauts peuvent créer des propriétés sans précédent, a déclaré Gavini. "Pourquoi ces défauts se forment-ils ? Comment pouvons-nous exploiter ces défauts pour provoquer des propriétés souhaitables plutôt que indésirables ? Dans des recherches précédentes, nous avons exploré l'énergie des dislocations individuelles dans le magnésium en vrac. Dans cette étude, nous étudions. Le résultat est l'image la plus détaillée de cette structure à ce jour, avec une précision quasi quantique. Gavini espère appliquer ces méthodes à un large éventail d'études.
"Si nous pouvons effectuer ces calculs à grande échelle avec une précision quasi quantique, cela signifie que nous pouvons concevoir de meilleurs matériaux grâce à la conception informatique, explorer des composés pour la découverte de médicaments et comprendre les nanoparticules et les systèmes de matériaux à de nouveaux niveaux. Détails de la fonctionnalité", Gavini dit. "Sans le calcul exascale et Frontier, nous ne serions pas en mesure d'effectuer ce type de calculs. Maintenant que nous savons comment le faire, nous pouvons largement appliquer ces méthodes pour explorer d'autres problèmes." peut être largement utilisé dans de nombreux domaines scientifiques et peut résoudre certains problèmes difficiles de longue date dans des domaines allant de l'aérospatiale à la médecine
Contenu de référence :
https://awards.acm.org/bell
https://news.engin. .umich.edu/2023/11/material-simulation-with-quantum-accuracy-wins-gordon-bell-prize/https://www.hpcwire.com/off-the-wire /ornls-frontier-achieves- la précision quasi quantique dans la simulation d'alliages en lice pour le prix gordon-bell/
Ce qui précède est le contenu détaillé de. pour plus d'informations, suivez d'autres articles connexes sur le site Web de PHP en chinois!
Articles Liés
Voir plus- L'intelligence artificielle donne un nouvel élan à l'industrie pharmaceutique chinoise
- La demande de puissance de calcul de l'IA a fortement augmenté et Shanghai Lingang construira une industrie de puissance de calcul à l'échelle de plusieurs dizaines de milliards.
- Lightning News | JD.com lance le grand modèle Yanxi AI pour les scénarios de vente au détail, médicaux, logistiques et autres
- Baidu lance le premier modèle médical « de niveau industriel » de Chine « Modèle de médecine spirituelle » : Baidu lance le premier modèle médical « de niveau industriel » de Chine « Modèle de médecine spirituelle »
- Le premier Forum du Sommet sur le développement collaboratif de la chaîne industrielle des robots du delta du fleuve Yangtze 2023 s'est tenu avec succès à Wuhu, Anhui.