
Maison >Périphériques technologiques >IA >Implémentation d'une simulation atomique basée sur la charge à l'aide d'un réseau neuronal à usage général pré-entraîné CHGNet
Implémentation d'une simulation atomique basée sur la charge à l'aide d'un réseau neuronal à usage général pré-entraîné CHGNet

- PHPzavant
- 2023-09-27 17:45:01724parcourir
Réécrit comme : Violet
Les simulations à grande échelle d'interactions électroniques complexes restent l'un des plus grands défis de la modélisation atomique. Alors que les champs de force classiques ne parviennent souvent pas à décrire les couplages entre les états électroniques et les réarrangements ioniques, une dynamique moléculaire ab initio plus précise souffre d'une complexité informatique qui empêche les simulations à longue et à grande échelle, ce qui est pertinent pour les techniques de recherche. Le phénomène est crucial
Récemment, des chercheurs de l'Université de Californie à Berkeley et du Lawrence Berkeley National Laboratory ont proposé un modèle de potentiel interatomique d'apprentissage automatique (MLIP) basé sur des réseaux de neurones graphiques : Crystal Hamiltonian Graph Neural Network (Crystal Hamiltonian Graph Neural Network (CHGNet), qui peut modéliser l'énergie potentielle universelle surfaces.
La recherche met en évidence l'importance des informations sur la charge pour capturer les réactions chimiques appropriées et fournit des informations sur les systèmes ioniques avec des degrés de liberté électroniques supplémentaires non observables avec les MLIP précédents.
La recherche était intitulée « CHGNet en tant que potentiel de réseau neuronal universel pré-entraîné pour la modélisation atomistique informée par la charge » et a été publiée dans « Nature Machine Intelligence » le 14 septembre 2023.

Les simulations à grande échelle, telles que la dynamique moléculaire (MD), sont des outils importants pour l'exploration informatique des matériaux à l'état solide. Cependant, modéliser avec précision les interactions électroniques et leurs effets subtils dans les simulations de dynamique moléculaire reste un défi de taille. Les méthodes empiriques telles que les champs de force classiques ne sont souvent pas assez précises pour capturer des interactions électroniques complexes.
La dynamique moléculaire ab initio (AIMD) combinée à la théorie fonctionnelle de la densité (DFT) peut être utilisée pour calculer explicitement les électrons dans la structure d'approximation fonctionnelle de la densité, produisant ainsi des valeurs élevées. -des résultats de fidélité avec une précision mécanique quantique. Les simulations AIMD polarisées en spin à long terme et à grande échelle, essentielles pour étudier la migration des ions, les transitions de phase et les réactions chimiques, sont difficiles et coûteuses en termes de calcul.
Les MLIP tels que ænet et DeepMD offrent des solutions prometteuses pour combler le fossé entre les méthodes de structure électronique coûteuses et les potentiels interatomiques classiques efficaces. Cependant, intégrer les effets importants de la valence sur la liaison chimique reste un défi dans MLIP.
La charge peut être représentée de nombreuses façons, depuis de simples étiquettes d'état d'oxydation jusqu'aux fonctions d'onde continue dérivées de la mécanique quantique. Le défi de l'intégration des informations de charge dans MLIP vient de nombreux facteurs, tels que l'ambiguïté de la représentation, la complexité de l'interprétation, la rareté des étiquettes, etc.
Ce qui doit être réécrit est : l'architecture CHGNet
CHGNet basée sur le projet matériel L'ensemble de données de trajectoire (MPtrj) est pré-entraîné sur l'énergie, la force, la contrainte et le moment magnétique. L'ensemble de données contient plus de 10 ans de calculs de théorie fonctionnelle de la densité sur 1,5 million de structures inorganiques. En incluant explicitement les moments magnétiques, CHGNet est capable d'apprendre et de représenter avec précision l'occupation orbitale des électrons, améliorant ainsi sa capacité à décrire les degrés de liberté atomiques et électroniques : Illustration : La distribution des éléments dans l'ensemble de données MPtrj. (Source : article) Ici, les chercheurs définissent la charge comme une propriété atomique (charge atomique) qui peut être déduite en incluant les moments magnétiques (magmoms). La recherche montre qu'en incorporant explicitement des magmoms spécifiques au site comme contraintes d'état de charge dans CHGNet, la régularisation de l'espace latent peut être améliorée et les interactions électroniques peuvent être capturées avec précision.
La fondation de CHGNet est GNN, où des couches convolutives graphiques sont utilisées. un ensemble de nœuds {vi} reliés par des arêtes {eij}. L'invariance de translation, de rotation et d'alignement est préservée dans GNN. CHGNet prend en entrée une structure cristalline avec des charges atomiques inconnues et génère les énergies, forces, contraintes et magmoms correspondants. Les structures décorées de charges peuvent être déduites des magmomes de champ et de la théorie des orbitales atomiques.

Le contenu réécrit est le suivant : Illustration : Architecture du modèle CHGNet. (Source : papier)
Dans CHGNet, les structures cristallines périodiques sont converties en graphiques atomiques  en recherchant les atomes voisins vj à l'intérieur de
en recherchant les atomes voisins vj à l'intérieur de  de chaque atome vi dans l'unité d'origine.
de chaque atome vi dans l'unité d'origine.
Contrairement à d'autres GNN, où les caractéristiques atomiques mises à jour 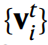 après t couches convolutives sont utilisées directement pour prédire l'énergie, CHGNet régularise les caractéristiques des nœuds
après t couches convolutives sont utilisées directement pour prédire l'énergie, CHGNet régularise les caractéristiques des nœuds
 après t−1 couches convolutives pour inclure des informations sur le magma. Les fonctionnalités régularisées
après t−1 couches convolutives pour inclure des informations sur le magma. Les fonctionnalités régularisées  contiennent des informations riches sur l'environnement ionique local et la distribution des charges. Par conséquent, la signature atomique
contiennent des informations riches sur l'environnement ionique local et la distribution des charges. Par conséquent, la signature atomique  utilisée pour prédire l’énergie, la force et la contrainte est la charge contrainte par des informations sur son état de charge. Par conséquent, CHGNet peut fournir des informations sur l’état de charge en utilisant uniquement les positions nucléaires et les identités atomiques comme entrées, permettant ainsi l’étude de la distribution des charges dans la modélisation atomique.
utilisée pour prédire l’énergie, la force et la contrainte est la charge contrainte par des informations sur son état de charge. Par conséquent, CHGNet peut fournir des informations sur l’état de charge en utilisant uniquement les positions nucléaires et les identités atomiques comme entrées, permettant ainsi l’étude de la distribution des charges dans la modélisation atomique.
Applications de CHGNet dans les matériaux à l'état solide
Les chercheurs ont démontré plusieurs applications de CHGNet dans les matériaux à l'état solide. Démontre les contraintes de charge et la régularisation de l'espace latent des charges atomiques dans Na2V2(PO4)3, et démontre le transfert de charge et la transition de phase de CHGNet dans LixMnO2, l'entropie électronique dans le diagramme de phase LixFePO4 et le conducteur superion au lithium de type grenat Li3+ Lithium (Li) et diffusivité dans xLa3Te2O12.
Pour rationaliser le traitement des charges atomiques, le matériau de cathode ionique sodium de type NASICON Na4V2(PO4)3 est utilisé comme exemple illustratif. En plus d'apprendre de la coordination spatiale des noyaux V, CHGNet distingue avec succès les ions V en deux groupes, V trivalent et V tétravalent, sans aucune connaissance préalable de la distribution de charge des ions V.

La figure montre la normalisation du moment magnétique et de l'espace caché dans Na2V2(PO4)3. (Cité de l'article)
L'étude de LixFePO4 a mis en évidence la capacité de CHGNet à distinguer

, ce qui est crucial pour inclure l'entropie électronique et la stabilité de phase à température finie.

Illustration : diagramme de phase LixFePO4 de CHGNet. (Source : article)
Dans l'étude de LiMnO2, il a été démontré que CHGNet peut mieux comprendre la relation entre la disproportion de charge et la transition de phase dans les systèmes d'oxydes de métaux de transition hétérovalents grâce aux informations de charge à long terme MD.
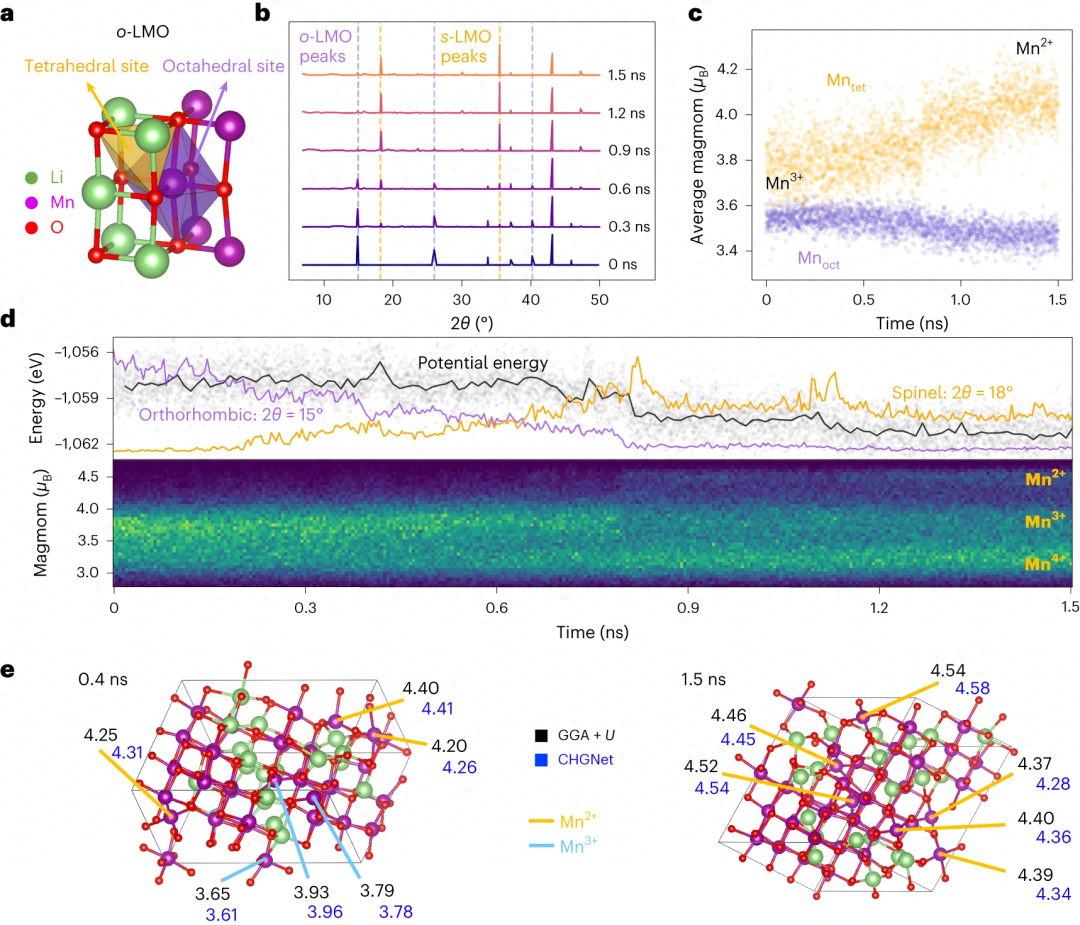
Contenu réécrit : Illustration : Transition de phase et différenciation de charge de Li0.5MnO2. (Citation de : article)
Ensuite, nous avons étudié la précision de CHGNet dans les simulations de dynamique moléculaire générale. Nous étudions la diffusion du lithium dans les conducteurs du grenat

Illustration : Diffusivité du lithium dans le grenat Li3La3Te2O12. (Source : article)
Les résultats montrent que CHGNet peut non seulement capturer avec précision l'effet du réseau de diffusion d'activation, mais que son énergie d'activation est également très cohérente avec les résultats DFT. Cela prouve que CHGNet peut capturer avec précision la forte interaction des ions lithium dans l’environnement local et a la capacité de simuler un comportement de diffusion hautement non linéaire. De plus, CHGNet est capable de réduire considérablement l'erreur dans les taux de diffusion simulés, et en s'étendant aux simulations nanosecondes, il est possible d'étudier des systèmes avec de faibles taux de diffusion
Peut être encore amélioré
Bien que les progrès ci-dessus aient été réalisés, il il y a encore place à l'amélioration Espace
Tout d'abord, l'utilisation de magmom pour l'inférence de valence ne garantit pas strictement la neutralité de la charge globale
Deuxièmement, bien que magmom soit une bonne heuristique pour les calculs de charge atomique pour la polarisation de spin dans les systèmes ioniques, on se rend compte que l'inférence de charge atomique pour les ions non magnétiques peut être ambiguë, donc une connaissance supplémentaire du domaine. Par conséquent, pour les ions sans magmom, le magmom centré sur l'atome ne peut pas refléter avec précision leur charge atomique, CHGNet déduira la charge de l'environnement, similaire à la fonctionnalité d'autres MLIP
Le modèle peut être encore amélioré en incorporant d'autres méthodes de représentation de charge, par exemple, fonctions de positionnement des électrons, polarisation électrique et partitionnement basés sur les orbitales atomiques. Ces méthodes peuvent être utilisées pour l'ingénierie des caractéristiques atomiques dans l'espace latent
CHGNet permet des simulations atomiques basées sur la charge, adaptées aux simulations informatiques à grande échelle pour étudier les systèmes hétérovalents, élargissant ainsi la portée de la chimie informatique, de la physique, de la biologie et de la science des matériaux. sur les phénomènes de couplage par transfert de charge
Veuillez cliquer sur le lien suivant pour consulter l'article : https://www.nature.com/articles/s42256-023-00716-3
Ce qui précède est le contenu détaillé de. pour plus d'informations, suivez d'autres articles connexes sur le site Web de PHP en chinois!
Articles Liés
Voir plus- Théorie de base de Java et classification des langages de programmation
- Présenté dans la principale publication internationale PNAS ! À partir d'ordinateurs théoriques, les scientifiques ont proposé un modèle de conscience - la « machine de Turing consciente »
- Théorie, implémentation et réglage des hyperparamètres des arbres de décision et des forêts aléatoires