
Maison >Périphériques technologiques >IA >Exploration et application de modèles de prédiction de la conformation du génome et de méthodes de criblage génétique informatique à haut débit
Exploration et application de modèles de prédiction de la conformation du génome et de méthodes de criblage génétique informatique à haut débit

- PHPzavant
- 2023-05-08 14:16:08887parcourir

Figure 0
Les différences de conformation du génome dans différents types de cellules déterminent la spécificité de l'expression des gènes, qui à son tour détermine les différences fonctionnelles dans différents types de cellules. Pendant longtemps, les méthodes expérimentales de détection de la conformation du génome, de l'hybridation in situ à la détection à haut débit telle que les technologies Hi-C et micro-C, sont généralement longues, laborieuses, coûteuses et présentent de fortes limitations techniques. Ces méthodes limitent considérablement l'application généralisée de ces techniques expérimentales dans le domaine de la recherche sur la conformation du génome, notamment dans l'étude de types de cellules rares et la nécessité de vérifier la relation causale entre la régulation de la conformation du génome à grande échelle. Les limites de ces méthodes ont également limité pendant longtemps les nouvelles découvertes dans le domaine de la régulation conformationnelle du génome tridimensionnel.

Figure 1
9 janvier 2023, Le laboratoire Aristotelis Tsirigos et le Broad Institute du MIT et de Harvard, NYU Grossman School of Medicine, le laboratoire de Xia Bo ont collaboré pour publier un article "Spécifique au type de cellule la prédiction de l'organisation de la chromatine 3D permet un criblage génétique in silico à haut débit" dans Nature Biotechnology.

Adresse papier : https://www.nature.com/articles/s41587-022-01612-8
Dans cette étude, Ph.D., New York University School de médecine Sheng Tan Jimin et le Dr Xia Bo ont d'abord proposé un nouveau modèle d'apprentissage automatique multimodal C.Origami pour prédire la conformation de la chromatine de types de cellules spécifiques, et ont proposé un nouveau criblage génétique informatique à haut débit (criblage génétique in silico) basé sur sur le principe du dépistage génétique. , ISGS), utilisée pour identifier les éléments génomiques fonctionnels spécifiques d'un type cellulaire et aider à découvrir de nouveaux mécanismes de régulation de la conformation de la chromatine.
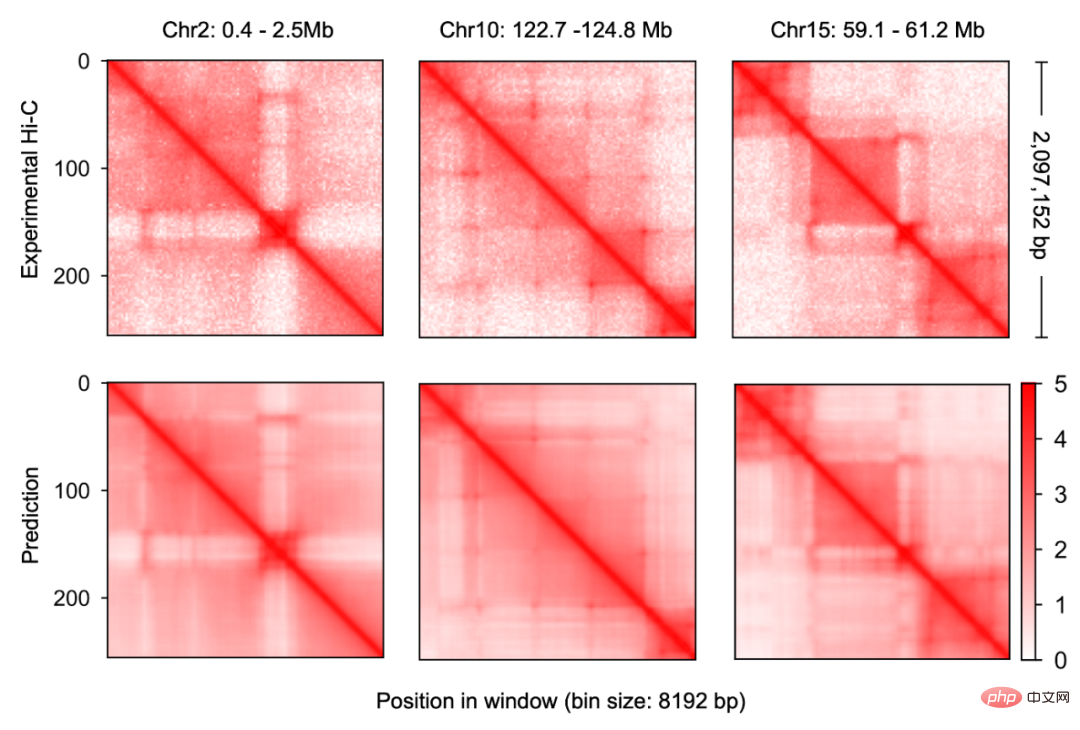
Figure 2
Les chercheurs ont d'abord construit un nouveau cadre d'apprentissage profond multimodal pour les données génomiques, Origami, afin qu'il puisse intégrer efficacement les informations sur les séquences d'ADN et les cellules génomiques fonctionnelles spécifiques informations, puis prédire de nouvelles informations génomiques. Grâce à un débogage répété et à une formation de modèles, les chercheurs ont découvert que l'intégration de la séquence d'ADN, de l'état de liaison du CTCF (CTCF ChIP-seq) et des signaux ATAC-seq comme informations d'entrée permet de prédire avec précision la conformation de la chromatine et d'utiliser la matrice bidimensionnelle Hi-C comme Prédisez l’objectif de sortie (Figure 1-2). Les informations d'entrée étaient de 2 millions de paires de bases d'ADN, CTCF ChIP-seq et ATAC-seq. Les chercheurs utilisent le codage Onehot pour coder des séquences d'ADN discrètes, tandis que CTCF ChIP-seq et ATAC-seq codent des caractéristiques non discrètes.

C.Le modèle Origami est divisé en trois parties, l'encodeur qui traite et compresse les informations sur l'ADN et le génome, la couche intermédiaire du transformateur et le décodeur Hi-C de sortie#🎜 🎜 #. L'encodeur se compose d'une série de ResNet 1D et de convolution striée pour encoder et compresser les informations d'entrée de 2 millions de paires de bases. À la fin de l'encodeur, le message d'une longueur de 2 millions est compressé à une longueur de 256 et utilisé comme message d'entrée pour le transformateur. Le mécanisme d'auto-attention de Transformer peut gérer l'interdépendance entre différentes régions génomiques et améliorer les performances globales du modèle. La matrice d'attention dans Transformer peut également améliorer l'interprétabilité du modèle. Les chercheurs ont converti le poids de l'attention en un « score d'attention » pour mesurer l'accent mis par le modèle sur différents domaines lors de la prédiction. Enfin, les chercheurs ont converti la sortie 1D du module Transformer en une matrice de contact/adjacence 2D en utilisant la « concaténation externe », qui a été utilisée comme information d'entrée pour le décodeur Hi-C. Le décodeur est un ResNet 2D dilaté. Les chercheurs ont ajusté les facteurs de dilatation des différentes couches afin que le champ récepteur à chaque position de pixel de la couche finale puisse couvrir toutes les informations d'entrée.
Ce modèle de prédiction de la conformation de la chromatine s'appelle C.Origami. Les chercheurs appellent C.Origami le premier modèle d’apprentissage profond multimodal en génomique. En raison de sa nature multimodale, C.Origami est capable de prédire avec précision (prédiction de novo) la conformation de la chromatine dans de nouveaux types de cellules qui n’y ont jamais été exposés. Par exemple, un modèle formé sur des cellules IMR-90 (fibroblastes pulmonaires) a pu prédire avec précision les conformations spécifiques de la chromatine dans les cellules GM12878 (lymphocytes B) (Figure 3).

Photo 3#🎜🎜 #
Les variantes structurelles - telles que les translocations chromosomiques - sont très courantes dans les tumeurs et modifient souvent les modèles d'interaction chromatine, ce qui peut affecter l'expression d'oncogènes ou de gènes suppresseurs de tumeurs. L'étude des effets de ces variations structurelles sur la conformation de la chromatine et l'expression des gènes est importante pour comprendre les mécanismes d'apparition et de progression des tumeurs. Ce type de recherche nécessite généralement l'utilisation d'expériences telles que 4C-seq ou Hi-C pour analyser la conformation chromatine des sites de variation structurelle, mais est souvent limitée par les ressources et le temps et est difficile à mener à grande échelle.Dans cette étude, C. Origami peut simuler des variations de séquences d'ADN dans des variables d'entrée, puis prédire de nouvelles interactions chromatiniennes dans le génome du cancer muté. Des études antérieures ont révélé que le modèle cellulaire CUTLL1 de leucémie lymphoblastique aiguë à cellules T (T-ALL) présentait une translocation chromosomique chr7-chr9 (Figure 4). En simulant informatiquement les variantes de translocation chromosomique, C. Origami a prédit avec précision la nouvelle structure TAD au niveau du site variante et a détecté une structure de « bande de chromatine » s'étendant de chr9 à chr7 (Figure 4).

Photo 4#🎜🎜 # Compte tenu de l'effet de prédiction précis de C.Origami et inspirés par le principe du dépistage génétique inverse, les chercheurs ont proposé une nouvelle méthode de dépistage génétique informatique à haut débit (criblage génétique in silico, ISGS) pour identifier systématiquement le type de cellules. -des éléments génomiques fonctionnels spécifiques et facilitent la découverte de nouvelles molécules régulatrices de coloration (Figure 5). Les chercheurs ont développé un cadre de criblage génétique informatique ISGS basé sur le modèle C. Origami pour l'identification systématique des éléments cis-régulateurs requis pour la conformation de la chromatine. En utilisant l’ISGS d’une résolution de 1 Ko à l’échelle du génome, les auteurs ont isolé des éléments cis-régulateurs (environ 1 % du génome) qui ont des effets importants sur la conformation de la chromatine. Ces séquences régulatrices de conformation de la chromatine présentent une dépendance différentielle sur la liaison CTCF et les signaux ATAC-seq (Fig. 5).

ISGS permet le criblage à haut débit des conformations de chromatine spécifiques à une cellule ou à une maladie. Les chercheurs ont réalisé l'ISGS dans les cellules CUTLL1, Jurkat et T normales et ont découvert qu'un élément cis-régulateur (CHD4-insu) proche du gène CHD4 était spécifiquement perdu dans les cellules T-ALL. Les résultats du dépistage indiquent que la perte isolante de CHD4-insu dans les cellules T-ALL pourrait permettre au gène CHD4 d'établir de nouvelles interactions chromatiniennes, régulant ainsi positivement l'expression de CHD4 et favorisant la prolifération des cellules leucémiques. ISGS peut également être utilisé pour découvrir systématiquement de nouveaux facteurs agissant en trans qui régulent la conformation de la chromatine. Grâce à l'analyse d'enrichissement d'importantes séquences régulatrices spécifiques au type de cellule et des sites de liaison aux facteurs de transcription, les chercheurs ont identifié les facteurs de régulation qui contribuent à la conformation du génome spécifique au type cellulaire. Il est intéressant de noter que des études antérieures ont montré que MAZ pouvait réguler la conformation de la chromatine avec le CTCF. Grâce à l'ISGS et à l'analyse de l'enrichissement en facteurs de transcription, les auteurs ont découvert que MAZ est considérablement enrichi dans les régions de chromatine ouvertes, tout en ne montrant qu'une faible liaison dans les régions de chromatine non ouvertes où se lie le CTCF. Ce résultat suggère que MAZ pourrait réguler la conformation du génome indépendamment du CTCF. Les chercheurs voient un grand potentiel dans les modèles d'apprentissage automatique multimodaux qui combinent la séquence d'ADN et les informations sur la chromatine dans la prédiction de la structure de la chromatine. L'architecture multimodale sous-jacente du modèle, Origami, peut être étendue à l'application d'autres données génomiques, telles que les modifications épigénétiques, l'expression des gènes, le criblage fonctionnel des mutations, etc. Les chercheurs prédisent que les futures recherches en génomique s’orienteront davantage vers l’utilisation de modèles d’apprentissage profond comme outils de dépistage génétique informatique primaire, complétés par une nouvelle génération de méthodes de recherche à haut débit validées par des expériences biologiques. Dans cette étude, Tan Jimin, doctorant à la faculté de médecine de l'Université de New York, est le premier auteur, et le Dr Aristotelis Tsirigos et le Dr Xia Bo sont les auteurs co-correspondants. Cette recherche a commencé avec le brainstorming de Xia Bo et Tan Jimin lors du confinement épidémique en octobre 2020. Après deux ans et demi d’amélioration et de peaufinage, elle a été officiellement publiée dans Nature Biotechnology en janvier 2023. Le code et les données de formation de ce projet ont été open source sur GitHub et Zenodo, et sont équipés de Google Colab pour une démonstration fonctionnelle. Adresse du projet : https://github.com/tanjimin/C.Origami Page d'accueil du laboratoire du Dr Xia Bo (Broad Institute du MIT et Harvard) : www.boxialab.org Tsirigos Lab (École de médecine Grossman de l'Université de New York) Page d'accueil : http://www.tsirigos.com
Ce qui précède est le contenu détaillé de. pour plus d'informations, suivez d'autres articles connexes sur le site Web de PHP en chinois!
Articles Liés
Voir plus- Tendances technologiques à surveiller en 2023
- Comment l'intelligence artificielle apporte un nouveau travail quotidien aux équipes des centres de données
- L'intelligence artificielle ou l'automatisation peuvent-elles résoudre le problème de la faible efficacité énergétique des bâtiments ?
- Co-fondateur d'OpenAI interviewé par Huang Renxun : les capacités de raisonnement de GPT-4 n'ont pas encore atteint les attentes
- Bing de Microsoft surpasse Google en termes de trafic de recherche grâce à la technologie OpenAI